Aufgabe 5 – Neurobiologie, Genetik
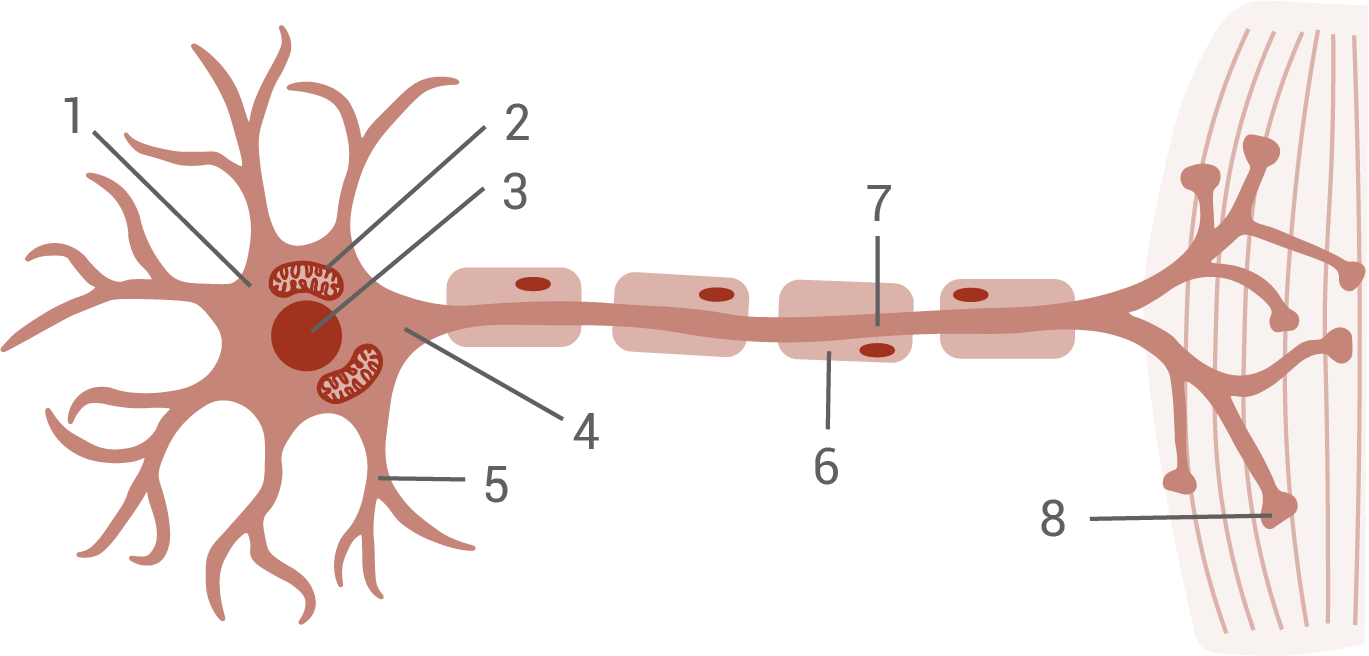
Motoneurone (Abbildung 1) sind Nervenzellen, die Aktionspotenziale zur Muskulatur leiten und diese zur Kontraktion anregen. Für die Funktion der Motoneuronen spielt unter anderem das „survival of motor neuron“-Protein (SMN-Protein) eine zentrale Rolle. Die Wirkungen des SMN-Proteins in Motoneuronen sind vielfältig. Fehlt das SMN-Protein, werden Motoneuronen geschädigt. Die daraus folgende, fehlende Aktivierung der Muskulatur führt letztlich zu deren Verkümmerung (Muskelatrophie).
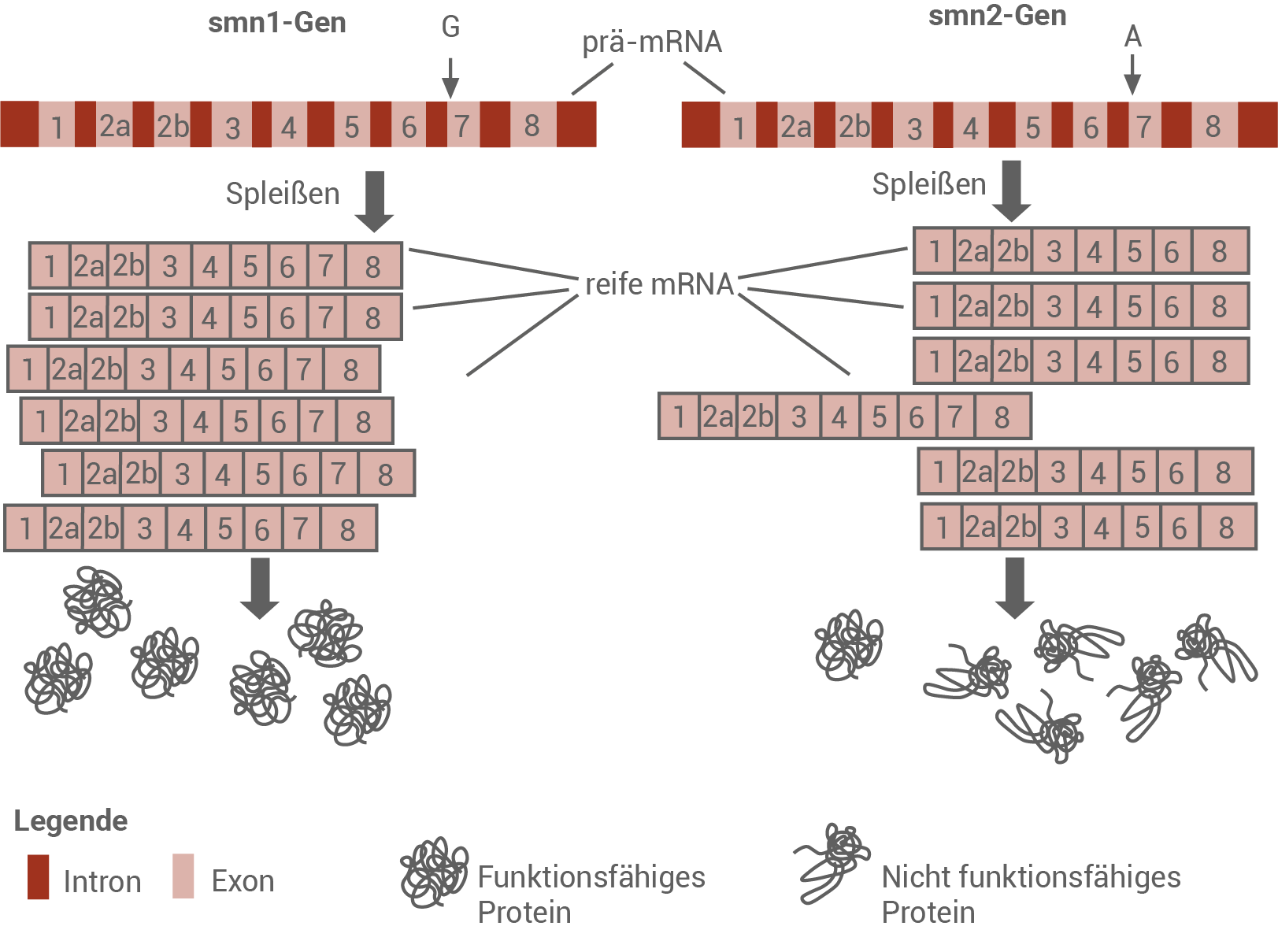
 Wie bei eukaryotischen Genen üblich, bestehen beide smn-Gene aus codierenden Abschnitten (Exons) und nicht-codierenden Abschnitten (lntrons). Nach der Transkription werden die lntrons aus der prä-mRNA herausgeschnitten und die Exons zur reifen mRNA zusammengefügt. Diesen Vorgang nennt man Spleißen. Die reife mRNA wird dann an den Ribosomen translatiert (Abb. 4).
Wie bei eukaryotischen Genen üblich, bestehen beide smn-Gene aus codierenden Abschnitten (Exons) und nicht-codierenden Abschnitten (lntrons). Nach der Transkription werden die lntrons aus der prä-mRNA herausgeschnitten und die Exons zur reifen mRNA zusammengefügt. Diesen Vorgang nennt man Spleißen. Die reife mRNA wird dann an den Ribosomen translatiert (Abb. 4).
 Eine gravierende Mutation im smn1-Gen kann zu Spinaler Muskelatrophie (SMA Typ 1) führen. Durch diese Mutation geht das gesamte Exon 7 des smn1-Gens verloren (Deletion). Die Mutation kann von Eltern auf ihre Kinder vererbt werden. Liegt die Mutation homozygot vor, kommt es bei den betroffenen Personen zum Krankheitsbild (rezessiver Erbgang), einer mehr oder weniger stark ausgeprägten Verkümmerung der Muskulatur. Die Schwere der Erkrankung ist abhängig von der Anzahl der im Genom der Patienten vorliegenden smn2-Genkopien. Manche Personen besitzen bis zu sechs Kopien dieses Gens.
Eine gravierende Mutation im smn1-Gen kann zu Spinaler Muskelatrophie (SMA Typ 1) führen. Durch diese Mutation geht das gesamte Exon 7 des smn1-Gens verloren (Deletion). Die Mutation kann von Eltern auf ihre Kinder vererbt werden. Liegt die Mutation homozygot vor, kommt es bei den betroffenen Personen zum Krankheitsbild (rezessiver Erbgang), einer mehr oder weniger stark ausgeprägten Verkümmerung der Muskulatur. Die Schwere der Erkrankung ist abhängig von der Anzahl der im Genom der Patienten vorliegenden smn2-Genkopien. Manche Personen besitzen bis zu sechs Kopien dieses Gens.

Abb. 1: Schematische Darstellung eines Motoneurons
1.1
Benenne die in Abbildung 1 mit Ziffern gekennzeichneten Strukturen eines Motoneurons.
2 VP
1.2
Zeichne ein Diagramm, das den Verlauf eines Aktionspotenzials zeigt (Größe ½ Seite) und erläutere die auf molekularer Ebene ablaufenden Prozesse, die zu diesem Spannungsverlauf führen.
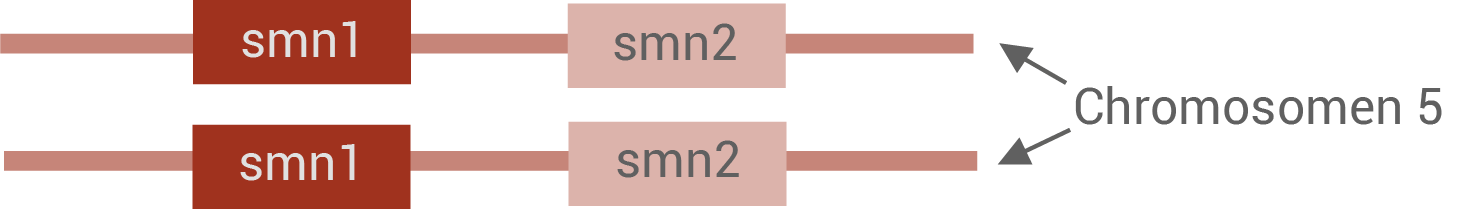
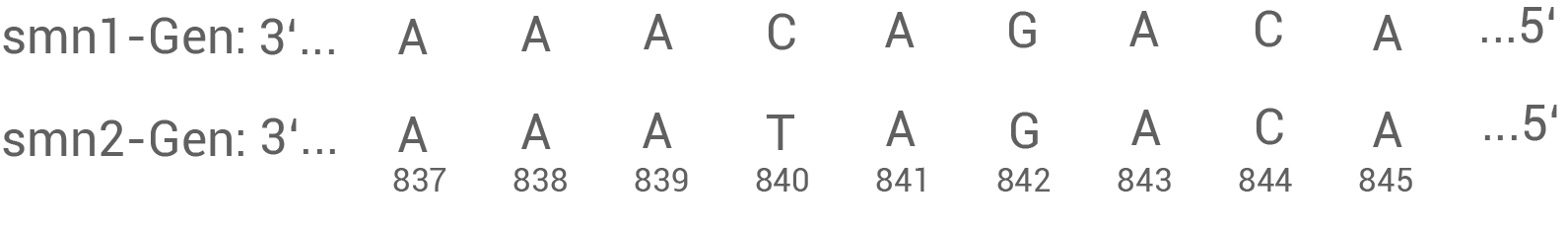
Das SMN-Protein aller Menschen wird von zwei verschiedenen Genen (smn1 und smn2) auf Chromosom 5 codiert (Abb. 2). Das smn2-Gen ist im Laufe der Evolution durch Duplikation (Verdopplung) des smn1-Gens entstanden und hat sich im weiteren Verlauf durch eine Punktmutation geringfügig verändert (Abb. 3).
4 VP
Abb. 2: Ausschnitt der homologen Chromosomen 5 mit smn1- und smn2-Genen bei gesunden Personen
Abb. 3: Punktmutation im Exon 7 des smn2-Gens (codogener Strang)
Abb. 4: Prozesse bei der Bildung von Genprodukten der Gene smn1 und smn2 bei gesunden Personen
2
Erläutere unter Verwendung der Abbildungen 3 und 4, weshalb die Deletion des Exons 7 in beiden smn1-Genen zur Erkrankung SMA Typ 1 führt und Personen mit einer erhöhten Anzahl an Duplikationen des smn2-Gens einen milderen Krankheitsverlauf aufweisen.
In einer Familie tritt bei einer Tochter ein schwerer Fall von Muskelatrophie auf. Um festzustellen, ob es sich dabei um SMA Typ 1 handelt, wird eine genetische Untersuchung vorgenommen. Hierzu werden zunächst die Exons 7 der smn1- und smn2-Gene der Tochter, der Eltern sowie einer gesunden Person durch Polymerasekettenreaktion vervielfältigt. Die vervielfältigte DNA wird mit Hilfe eines Restriktionsenzyms geschnitten. Die hierbei erhaltenen DNA-Fragmente werden im Anschluss mit einer Gelelektrophorese aufgetrennt (Abbildung 5).
4 VP
Abb. 5: Ergebnis der Gelelektrophorese (schematische Darstellung)
3.1
Erkläre das Zustandekommen des Bandenmusters bei einer gesunden Person.
3 VP
3.2
Übertrage die Abbildung 5 in Deine Reinschrift und ergänze die zu erwartenden Banden für die Proben der Familie für den Fall, dass es sich bei der Erkrankung der Tochter um den SMA Typ 1 handelt. Achte in Deiner Darstellung darauf, unterschiedliche DNA Mengen durch unterschiedlich starke Banden zu kennzeichnen. Begründe Deine Darstellung.
Spinale Muskelatrophie Typ 1 ist derzeit nicht heilbar. Der Krankheitsverlauf kann jedoch durch Gentherapie mithilfe des Medikaments Zolgensma® begünstigt werden. Bei Zolgensma® handelt es sich um gentechnisch veränderte Viren, die in ihrem Genom das intakte smn1-Gen integriert haben. Diese Viren werden dem Patienten mittels Infusion in die Blutbahn verabreicht. Die veränderte DNA der Viren gelangt so in Zellen der erkrankten Personen.
3 VP
4
Gib eine Erklärung dafür, dass diese Therapie zu einer Verbesserung der Symptome, nicht aber zu einer vollstandigen Heilung führt.
Bevor das Medikament verabreicht wird, muss untersucht werden, ob der Patient in der Vergangenheit bereits Kontakt mit dem verwendeten Virus hatte.
2 VP
5
Erkläre die Notwendigkeit dieser Maßnahme.
2 VP
20 VP
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?
1.1
Benennung der Strukturen:
- Zellsoma
- Mitochondrium
- Zellkern
- Axonhügel
- Dendrit
- Schwannsche Zelle (Myelin-/Markscheide)
- Axon
- Endknöpfchen (Synapse, motorische Endplatte)
1.2
Diagramm mit Aktionspotenzial
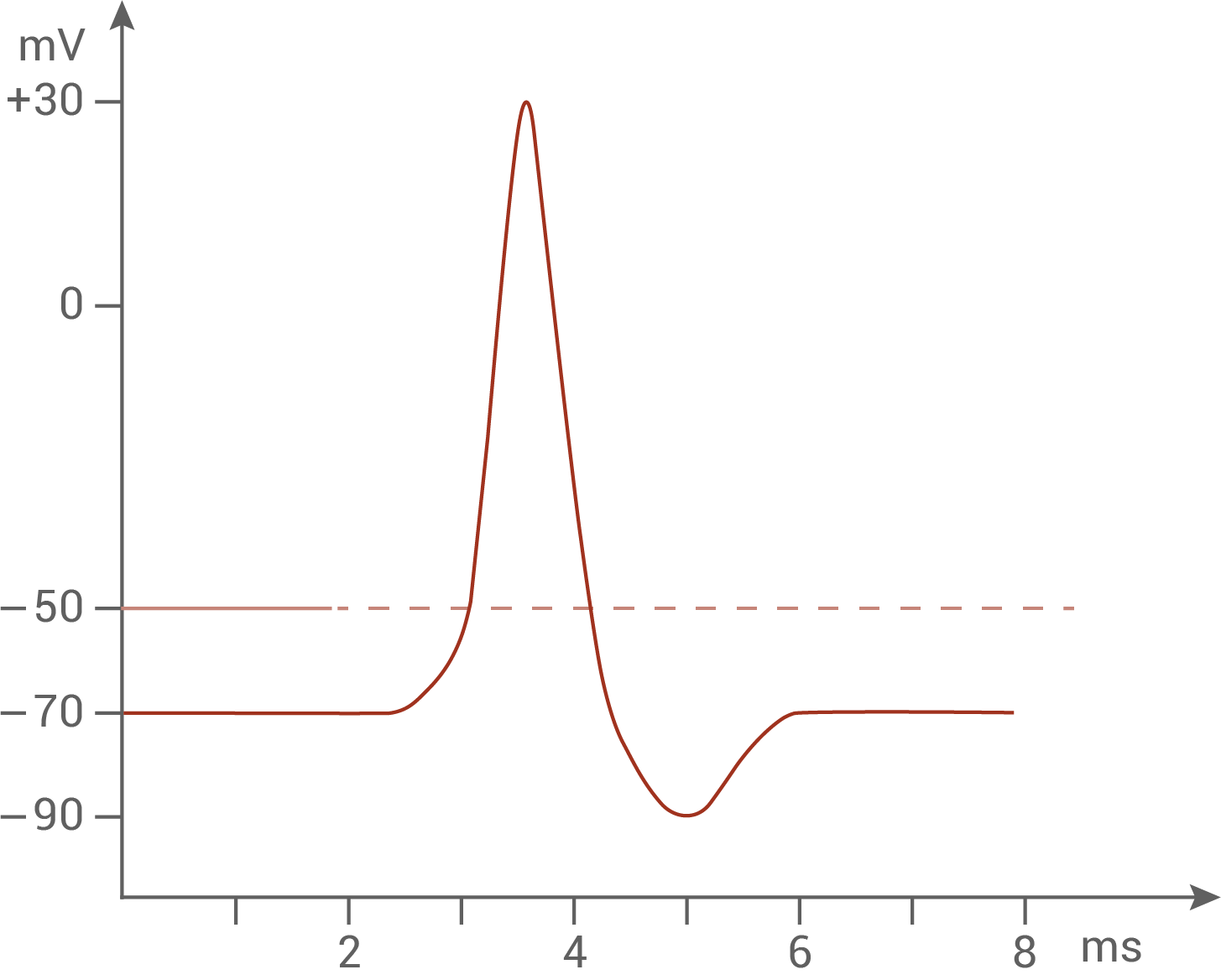 Mehrere elektrische Impulse gelangen über die Dendriten zum Axonhügel. Dort werden sie aufsummiert, bis der Schwellenwert von -50 mV durch eine Depolarisation der Membranspannung überschritten wird. Spannungsgesteuerte Natriumionenkanäle öffnen nun, und es gelangen Natriumionen ins Innere des Axons. Dieser Prozess führt zu einer Depolarisation der Membranspannung bis ca. + 30 mV (Spannungsumkehr). Die Natriumionenkanäle schließen, und kurz darauf öffnen die Kaliumionenkanäle. Es kommt zu einem Ausstrom von positiven Kaliumionen, was zu einer Repolarisation der Membran führt. Dabei strömen so lange Kaliumionen aus, bis das Membranpotenzial unter das Ruhepotenzial von -70 mV fällt. Die Kaliumionenkanäle schließen. Nach der Hyperpolarisation kann das Ruhepotenzial wieder hergestellt werden. Erst dann ist die Entstehung eines neuen Aktionspotenzials möglich.
Mehrere elektrische Impulse gelangen über die Dendriten zum Axonhügel. Dort werden sie aufsummiert, bis der Schwellenwert von -50 mV durch eine Depolarisation der Membranspannung überschritten wird. Spannungsgesteuerte Natriumionenkanäle öffnen nun, und es gelangen Natriumionen ins Innere des Axons. Dieser Prozess führt zu einer Depolarisation der Membranspannung bis ca. + 30 mV (Spannungsumkehr). Die Natriumionenkanäle schließen, und kurz darauf öffnen die Kaliumionenkanäle. Es kommt zu einem Ausstrom von positiven Kaliumionen, was zu einer Repolarisation der Membran führt. Dabei strömen so lange Kaliumionen aus, bis das Membranpotenzial unter das Ruhepotenzial von -70 mV fällt. Die Kaliumionenkanäle schließen. Nach der Hyperpolarisation kann das Ruhepotenzial wieder hergestellt werden. Erst dann ist die Entstehung eines neuen Aktionspotenzials möglich.
3.1
Erkrankung mit SMA Typ 1:
Die Deletion von Exon 7 im smn1-Gen führt zu einem Verlust der Funktionsfähigkeit des SMN-Proteins. Kommt es zu einer Mutation in beiden Allelen, entsteht kein funktionsfähiges SMN-Protein und es findet außerdem keine ausreichende Kompensation durch smn2-Gen statt. Dieser Prozess führt zu einer Schädigung der Motoneuronen und einem Abbau der durch die Neuronen innervierten Muskulatur.
Bei einem milden Verlauf findet eine teilweise Kompensation durch Genprodukte des smn2-Gens statt. Nur ein geringer Anteil der reifen mRNA-Transkripte des smn-2-Gens enthält noch das Exon 7. Es entstehen geringe Mengen eines funktionstüchtigen SMN-Proteins. Je mehr smn-2-Genkopien im Genom vorhanden ist, desto stärker fällt der Kompensationseffekt aus, und desto milder ist der Verlauf.
3.1
Bandenmuster einer gesunden Person:
DNA-Fragmente wandern aufgrund ihres negativ geladenen Phosphatrückgrat vom Minuspol zum Pluspol. Kurze Fragmente kommen dabei schneller durch die Maschen des Gels als längere Fragmente.
Das Bandenmuster einer gesunden Person kommt folgendermaßen zustande:
Exon 7 des smn-1-Gens ist vorhanden, somit gibt es auch keine Schnittstelle für das Restriktionsenzym. Es entsteht ein 200 bp langes Fragment. Dieses legt aufgrund seiner Länge die kürzeste Strecke im Gel zurück. Exon 7 des smn2-Gens ist ebenfalls vorhanden. Durch die Mutation ist eine Schnittstelle entstanden, an der das Restriktionsenzym ansetzen kann. Das Gen wird in ein 176 bp und ein 24 bp langes Fragment geschnitten, wobei das kürzeste Fragment die längste Strecke im Gel zurücklegen kann.
Exon 7 des smn-1-Gens ist vorhanden, somit gibt es auch keine Schnittstelle für das Restriktionsenzym. Es entsteht ein 200 bp langes Fragment. Dieses legt aufgrund seiner Länge die kürzeste Strecke im Gel zurück. Exon 7 des smn2-Gens ist ebenfalls vorhanden. Durch die Mutation ist eine Schnittstelle entstanden, an der das Restriktionsenzym ansetzen kann. Das Gen wird in ein 176 bp und ein 24 bp langes Fragment geschnitten, wobei das kürzeste Fragment die längste Strecke im Gel zurücklegen kann.
3.2
Bandenmuster einer SMA Typ 1 erkrankten Tochter:
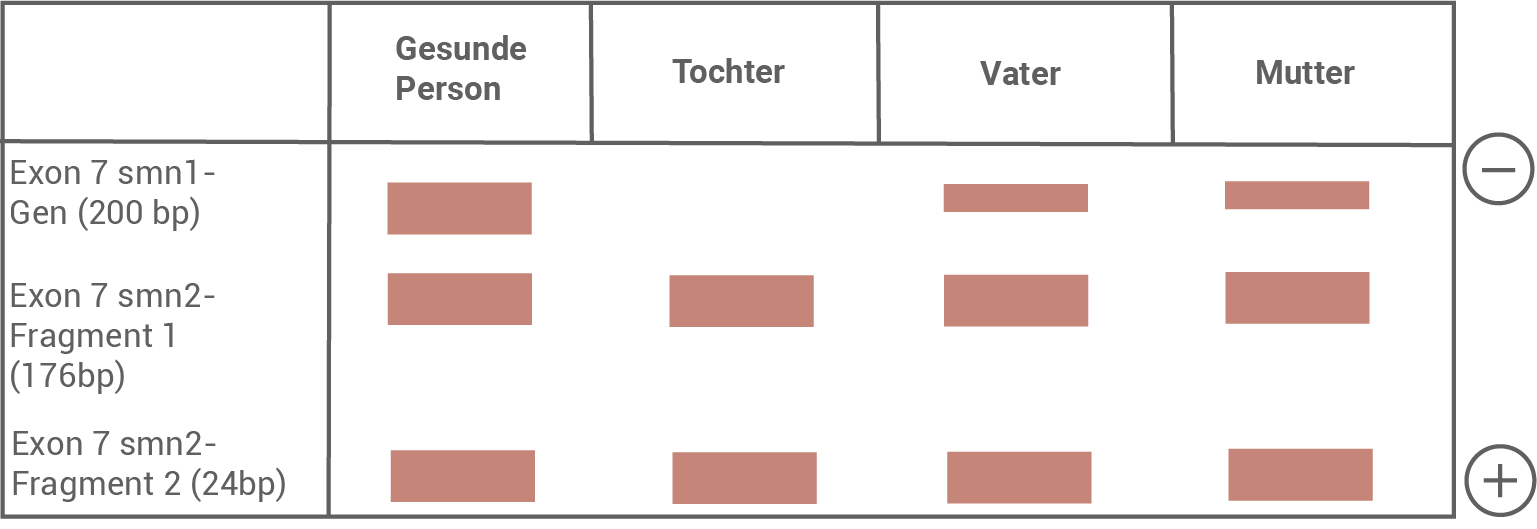 Wenn die Tochter an SMA Typ 1 erkrankt, dann fehlt das Exon 7 auf beiden homologen Chromosomen 5. Es gibt daher keine 200 bp langen DNA-Fragmente, und die entsprechende Bande fehlt vollständig. Da das smn2-Gen unverändert bleibt, entsprechen die beiden unteren Banden denen einer gesunden Person.
Wenn die Tochter an SMA Typ 1 erkrankt, dann fehlt das Exon 7 auf beiden homologen Chromosomen 5. Es gibt daher keine 200 bp langen DNA-Fragmente, und die entsprechende Bande fehlt vollständig. Da das smn2-Gen unverändert bleibt, entsprechen die beiden unteren Banden denen einer gesunden Person.
Bei einer erkrankten Tochter müssen beide (gesunden) Elternteile bezüglich des smn1-Gens heterozygot sein. Sowohl Vater als auch Mutter verfügen also nur über ein Chromosom mit Exon 7 im smn1-Gen. Auf dem homologen Chromosom fehlt Exon 7 im smn1-Gen. Jedes Elternteil hat eine Bande für die Fragmente mit 200 bp, diese ist jedoch nur halb so dick wie bei einer homozygot gesunden Person.
Bei einer erkrankten Tochter müssen beide (gesunden) Elternteile bezüglich des smn1-Gens heterozygot sein. Sowohl Vater als auch Mutter verfügen also nur über ein Chromosom mit Exon 7 im smn1-Gen. Auf dem homologen Chromosom fehlt Exon 7 im smn1-Gen. Jedes Elternteil hat eine Bande für die Fragmente mit 200 bp, diese ist jedoch nur halb so dick wie bei einer homozygot gesunden Person.
4
Gentherapie mittles Zolgensma®:
Durch das Einbringen der viralen DNA mit smn1-Gen in den Zellkern der Zellen des Patienten, wird vermehrt funktionstüchtiges SMN-Protein gebildet. Dies führt zu einer Verbesserung der Symptome. Eine vollständige Heilung ist aufgrund folgender Faktoren nicht möglich:
- Nicht alle (Nerven-)Zellen des Patienten nehmen das Virus auf.
- Die aufgenommene DNA wird mit der Zeit in der Zelle abgebaut.
- Zellen, die das smn-1-Gen aufgenommen haben, sterben nach einer Weile ab.
5
Warum der Patient auf Kontakt mit dem Virus untersucht werden sollte:
Durch eine mögliche frühere Infektion mit dem Virus sind durch die abgelaufene Immunreaktion spezifische Antikörper und entsprechende Gedächtniszellen vorhanden. Die Verabreichung des Impfstoffes entspricht einer erneuten Infektion und löst eine Sekundärantwort aus. Es kommt zu einer Antigen-Antikörper-Reaktion, und das Medikament wird unwirksam.