HT 2
Thema: Die Pelizaeus-Merzbacher-Krankheit (PMK)
1.
Ermittle den wahrscheinlichen Erbgang der PMK, indem du andere Erbgänge begründet ausschließt (Material A). Gib für diesen Erbgang die möglichen Genotypen der Personen 5, 6, 8, 17 und 20 an (Material A).
(10 Punkte)
2.
Gib die Aminosäuresequenzen für die in Tabelle 1 dargestellten DNA-Sequenzen an und leite den Mutationstyp und die Folgen für das PLP1-Protein ab (Materialien B und D). Erkläre die Methode der DNA-Gelelektrophorese. Werte die in Abbildung 2 gezeigten Ergebnisse mithilfe von Tabelle 1 aus und leite die Genotypen von Mutter und Sohn ab (Material B).
(26 Punkte)
3.
Erkläre die Bedeutung der Myelinscheiden für die Erregungsweiterleitung. Fasse die in Material C dargestellten Ergebnisse zusammen und erläutere diese im Hinblick auf die neurophysiologischen Auswirkungen der PLP1-Mutation bei Mäusen (Material C). Stelle auf dieser Basis eine Hypothese zur Erklärung der Symptome einer PLP1-Mutation beim Menschen auf (Materialien A und C).
(18 Punkte)
Material A: Vererbung der Pelizaeus-Merzbacher-Krankheit
Die Pelizaeus-Merzbacher-Krankheit, abgekürzt PMK, ist eine sehr seltene angeborene Erkrankung des Zentralnervensystems. Die Krankheit beginnt meist im Kindesalter und geht mit einer Vielzahl von Symptomen einschließlich Verzögerungen der geistigen und körperlichen Entwicklung sowie Lähmungen der Muskulatur einher. Abbildung 1 zeigt einen Ausschnitt aus dem Stammbaum einer von der PMK betroffenen Familie.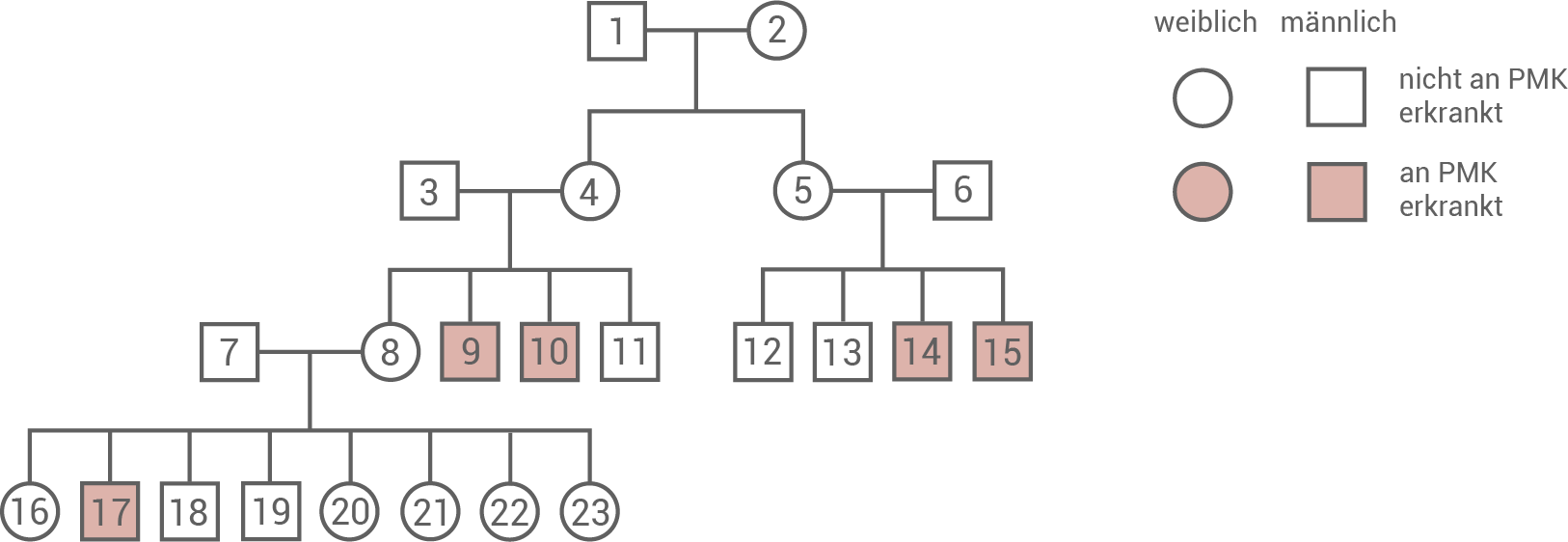
Abbildung 1  Stammbaum einer von der PMK betroffenen Familie
Stammbaum einer von der PMK betroffenen Familie
Material B: Genetische Ursachen der PMK
Ursache für die PMK sind Mutationen im PLP1-Gen, das für das Proteolipid-Protein 1 codiert. Es sind viele verschiedene Mutationen bekannt, welche die PMK auslösen. Bei einer betroffenen Familie wurde untersucht, welche genetische Ursache hier für die PMK verantwortlich war. Dafür wurde das PLP1-Allel eines männlichen Erkrankten sequenziert und mit der Sequenz des Normal-Allels des PLP1-Gens verglichen (Tabelle 1).
Tabelle 1 Sequenzausschnitt des PLP1-Gens. Es ist der nicht-codogene DNA-Strang dargestellt. Die Triplett-Nummern sind angegeben.

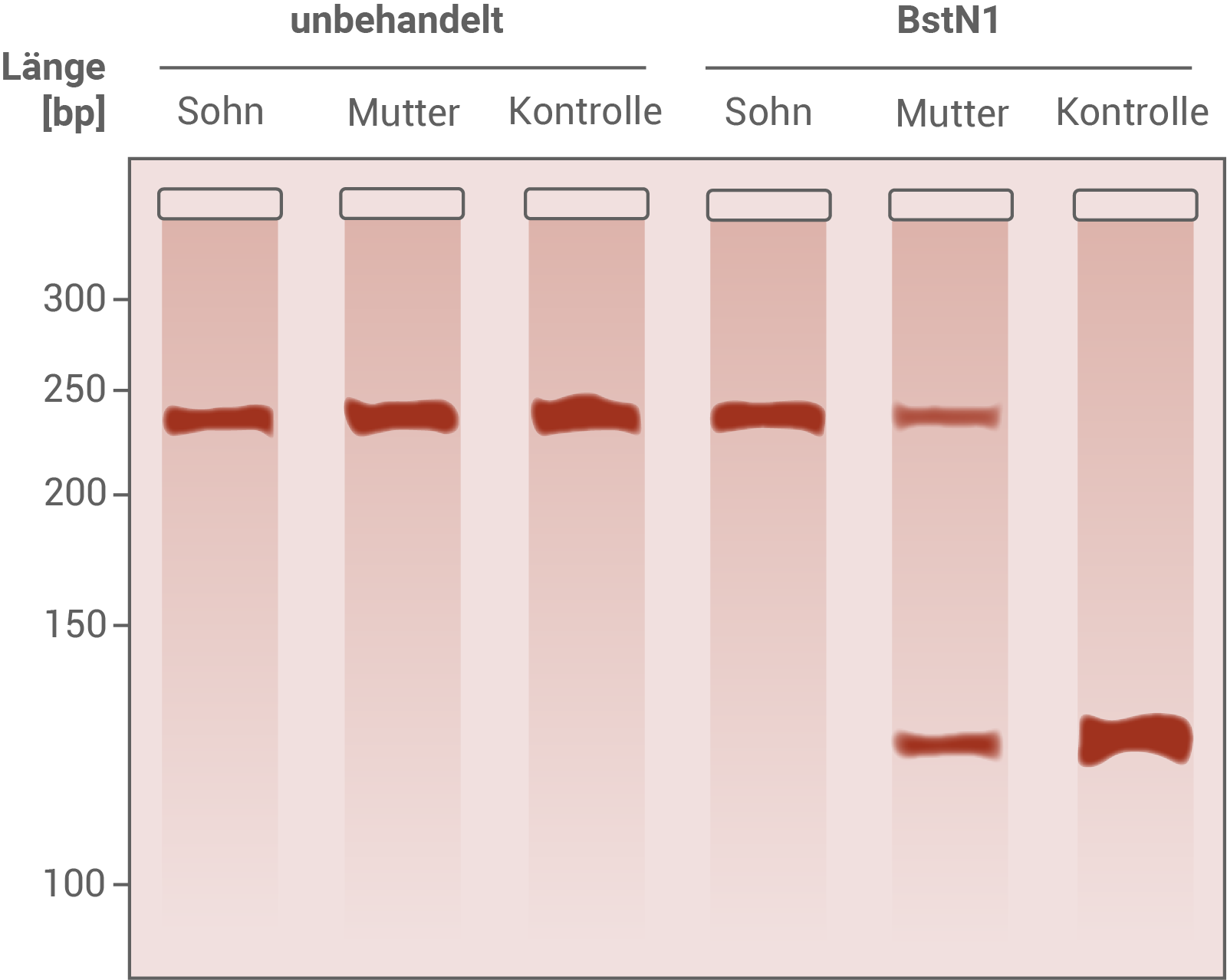
Abbildung 2 Ergebnis der Gelelektrophorese von unbehandelten und mit BstN1 behandelten PCR-Produkten des PLP1-Gens. bp: Basenpaare
Material C: Auswirkungen von PLP1-Mutationen
Die Aminosäure-Sequenz des PLP1-Proteins ist in Menschen und Mäusen identisch. Daher wurden Untersuchungen zu den neurophysiologischen Auswirkungen von PLP1-Mutationen an gentechnisch veränderten Mäusen durchgeführt. Diese Mäuse enthielten eine Mutation des PLP1-Gens, die bei Menschen zu einer besonders schweren Ausprägung der PMK führt. In Experimenten wurden diese transgenen Mäuse verschiedenen Alters mit jeweils gleich alten Wildtyp-Mäusen verglichen.
Die neurophysiologischen Untersuchungen erfolgten am Sehnerv. Der Sehnerv leitet elektrische Signale von den Lichtsinneszellen im Auge zum Gehirn. Im Sehnerv sind etwa eine Million Axone gebündelt. An einem Punkt des Sehnervs wurde der Anteil von myelinisierten Axonen an der Gesamtzahl der Axone bestimmt (Abbildung 3).
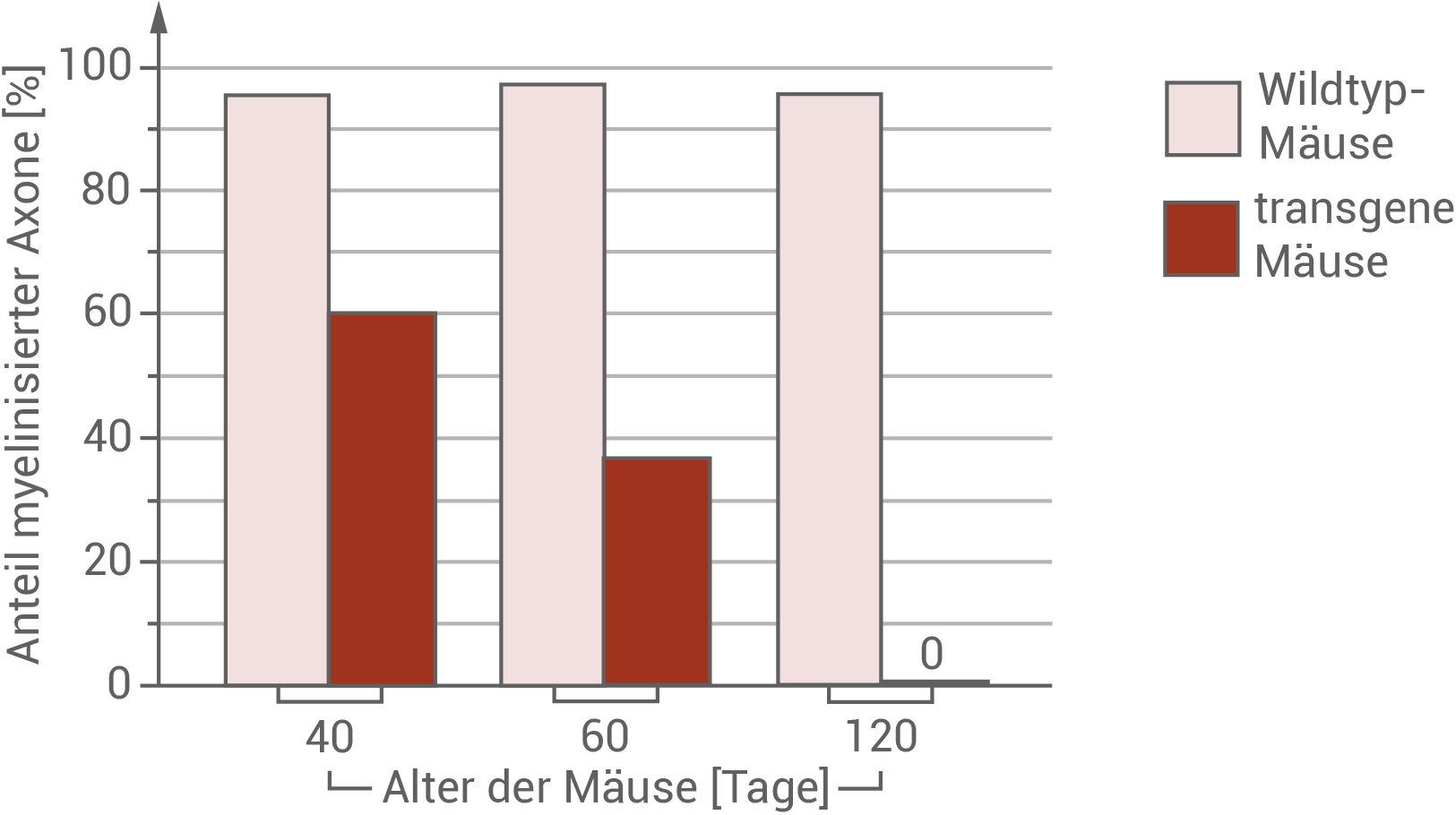
Abbildung 3 Anteil myelinisierter Axone im Sehnerv von Wildtyp-Mäusen und transgenen Mäusen mit PLP1-Mutation
Außerdem wurde die Erregungsleitung am Sehnerv untersucht. Dazu wurde die Dauer der Erregungsleitung zwischen Anfang und Ende des Sehnervs nach einem künstlichen Reiz gemessen (Abbildung 4).
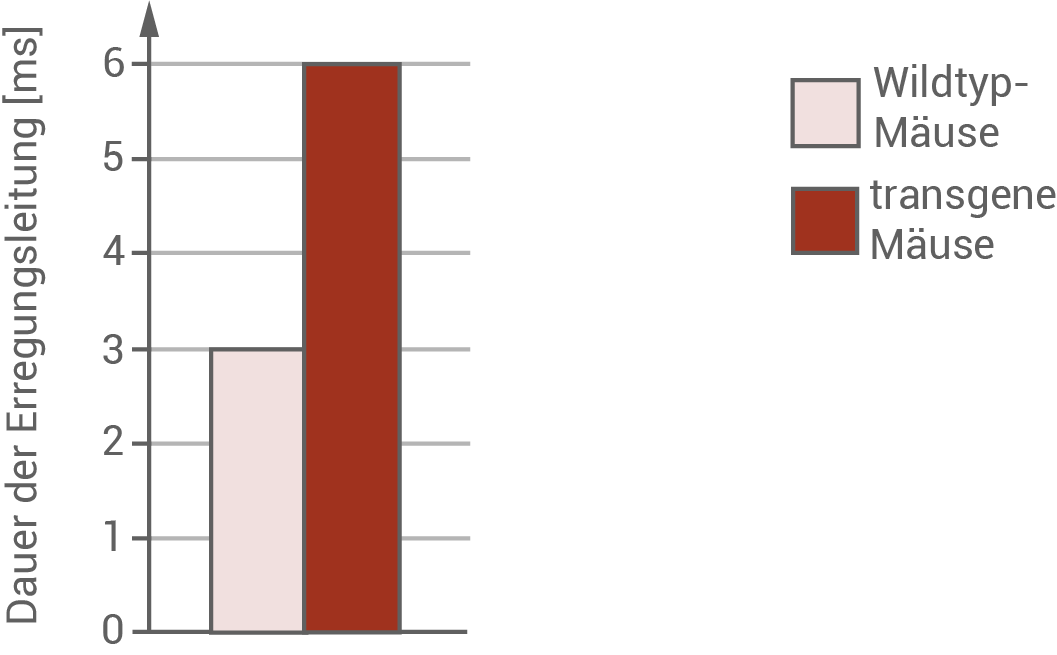
Abbildung 4 Dauer der Erregungsleitung am Sehnerv von 40 Tage alten Wildtyp-Mäusen und transgenen Mäusen mit PLP1-Mutation
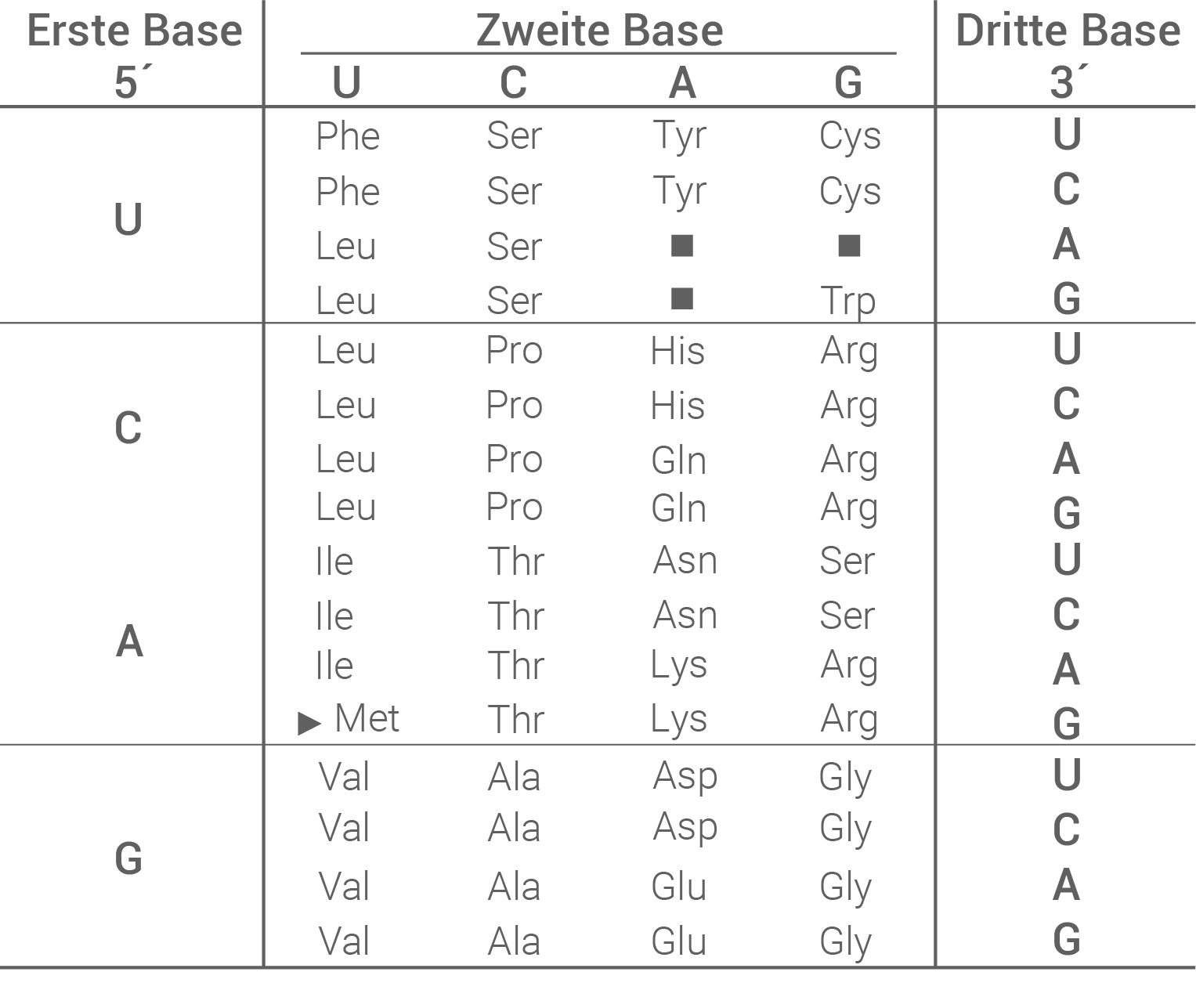
Material D: Codesonne und Tabelle zum genetischen Code

Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?
1.
Erbgang der PMK:
Die Analyse des Stammbaums zeigt, dass die PMK höchstwahrscheinlich X-chromosomal-rezessiv vererbt wird. Wäre der Erbgang dominant, so müsste mindestens ein Elternteil der erkrankten Personen ebenfalls die Krankheit ausprägen. Nur im Fall der rezessiven Vererbung können die Eltern ein betroffenes Allel an ihre Kinder vererben, die Krankheit selbst aber nicht ausprägen. In dem Stammbaum sind nur Männer von der Krankheit betroffen. Das spricht für einen X-chromosomalen Erbgang, da Männer nur ein X-Chromosom aufweisen. Wenn sie also ein betroffenes X-Chromosom vererbt bekommen, können sie es – im Gegensatz zu Frauen – nicht mit einem gesunden X-Chromosom kompensieren.
Genotypen:
Es sei X: Betroffenes X-Chromosom, x: Gesundes X-Chromosom und y: Gesundes Y-Chromosom.
- Person 5: Xx
- Person 6: xy
- Person 8: Xx
- Person 17: Xy
- Person 20: Xx oder xx
2.
Aminosäuresequenzen für die in Tabelle 1 dargestellten DNA-Sequenzen:
Normal-Allel
mRNA: 5' GCU UUC CCU GGC AAG GUU 3'
Aminosäuresequenz: Ala - Phe - Pro - Gly - Lys - Val mutiertes Allel
mRNA: 5' GCU UUC UCU GGC AAG GUU 3'
Aminosäuresequenz: Ala - Phe - Ser - Gly - Lys - Val Mutationstyp und Folgen für das PLP1-Protein: Im mutierten Allel tritt innerhalb der Nukleotidsequenz eine Punktmutation an Position 1 in Triplett 215 auf. Die Punktmutation ist eine Substitutionsmutation. Der Basenaustausch führt bei der Translation nicht zum Einbau von Prolin sondern zu Serin. Daher handelt es sich um eine Missense-Mutation. Der Basenaustausch kann zu veränderten chemische Wechselwirkungen mit den übrigen Aminosäuren in der Polypeptidkette führen. Möglicherweise führt das zu einer veränderten Faltung des Proteins. Das Protein verliert dadurch eventuell seine Funktion oder tritt mit anderen Stoffen in Interaktion als zuvor. Methode der DNA-Gelelektrophorese: Das Verfahren der Gelelektrophorese dient der Unterscheidung geladener Teilchen (wie zum Beispiel DNA-Fragmente) nach ihrer Größe. Durch eine anschließende Färbung des Gels können diese Fragmente sichtbar gemacht werden. Die Gelelektrophorese besteht aus einer Kammer, in der sich ein Agarose-Gel befindet. Das Gel ist strukturell wie ein engmaschiges Netz aufgebaut, wodurch größere Fragmente langsamer wandern als kleinere. Auf der Kathodenseite des Gels werden die zu untersuchenden Proben gemeinsam mit einem Ladepuffer in dafür vorgesehene Taschen im Gel gegeben. In der Regel wird in eine äußere Tasche ein Größenmarker gegeben, um die DNA-Fragmente später zuordnen zu können. Im Anschluss wird eine elektrische Spannung angelegt, und die Fragmente beginnen zu wandern. Da die DNA polar und durch ihr Phosphatrückgrat negativ geladen ist, wandert sie im Gel von der Kathode in Richtung Anode. Nach einer definierten Zeitspanne, wird das Gel von der Spannungsquelle getrennt, und mit einem Farbstoff eingefärbt, der die Banden im UV-Licht sichtbar macht. Dadurch und durch den Vergleich mit dem Größenmarker kann die Länge und Häufigkeit der erhaltenen DNA Fragmente ermittelt werden. Auswertung der in Abbildung 2 gezeigten Ergebnisse: Die unbehandelten DNA-Proben wurden wie erwartet nicht von einem Restriktionsenzym geschnitten, und weisen alle eine Länge von etwa 260 bp auf. Das Restriktionsenzym BstN1 scheidet in der Erkennungssequenz 5'-CCTGG-3'. Diese Sequenz liegt innerhalb des PLP1-Gens. Bei nicht-mutierten Allelen ist die Erkennungssequenz intakt, und das Enzym kann schneiden. Im mutierten Allel führt die Basensubstitution jedoch dazu, dass die Erkennungssequenz nicht mehr von BstN1 gefunden wird. Mutierte Allele werden also nicht geschnitten. Die Kontroll-DNA trägt keine Mutation auf dem PLP1-Gen in beiden Allelen. Daher wird die DNA-Probe hier an zwei Stellen geschnitten, und es entstehen zwei Fragmente gleicher Länge. Daher lässt sich in der Kontrolle eine dicke Bande mit einer Länge von etwa 130 bp erkennen. Die Mutter besitzt ebenfalls eine Bande mit der Länge von 130 bp, aber auch eine mit der Länge 260 bp. Daher konnte das Restriktionsenzym bei ihr nur in einem Allel schneiden, und sie besitzt ein gesundes und ein mutiertes Allel. Beide Banden sind im Vergleich zu den anderen Proben blass, da hier jeweils nur ein Teil der DNA vorliegt. Der Sohn zeigt nur eine Bande, die sich auf derselben Höhe befindet, wie bei unbehandelten Proben. Das bedeutet, dass der Sohn eine Mutation trägt, und das Restriktionsenzym nicht schneiden konnte. Genotypen von Mutter und Sohn:
mRNA: 5' GCU UUC CCU GGC AAG GUU 3'
Aminosäuresequenz: Ala - Phe - Pro - Gly - Lys - Val mutiertes Allel
mRNA: 5' GCU UUC UCU GGC AAG GUU 3'
Aminosäuresequenz: Ala - Phe - Ser - Gly - Lys - Val Mutationstyp und Folgen für das PLP1-Protein: Im mutierten Allel tritt innerhalb der Nukleotidsequenz eine Punktmutation an Position 1 in Triplett 215 auf. Die Punktmutation ist eine Substitutionsmutation. Der Basenaustausch führt bei der Translation nicht zum Einbau von Prolin sondern zu Serin. Daher handelt es sich um eine Missense-Mutation. Der Basenaustausch kann zu veränderten chemische Wechselwirkungen mit den übrigen Aminosäuren in der Polypeptidkette führen. Möglicherweise führt das zu einer veränderten Faltung des Proteins. Das Protein verliert dadurch eventuell seine Funktion oder tritt mit anderen Stoffen in Interaktion als zuvor. Methode der DNA-Gelelektrophorese: Das Verfahren der Gelelektrophorese dient der Unterscheidung geladener Teilchen (wie zum Beispiel DNA-Fragmente) nach ihrer Größe. Durch eine anschließende Färbung des Gels können diese Fragmente sichtbar gemacht werden. Die Gelelektrophorese besteht aus einer Kammer, in der sich ein Agarose-Gel befindet. Das Gel ist strukturell wie ein engmaschiges Netz aufgebaut, wodurch größere Fragmente langsamer wandern als kleinere. Auf der Kathodenseite des Gels werden die zu untersuchenden Proben gemeinsam mit einem Ladepuffer in dafür vorgesehene Taschen im Gel gegeben. In der Regel wird in eine äußere Tasche ein Größenmarker gegeben, um die DNA-Fragmente später zuordnen zu können. Im Anschluss wird eine elektrische Spannung angelegt, und die Fragmente beginnen zu wandern. Da die DNA polar und durch ihr Phosphatrückgrat negativ geladen ist, wandert sie im Gel von der Kathode in Richtung Anode. Nach einer definierten Zeitspanne, wird das Gel von der Spannungsquelle getrennt, und mit einem Farbstoff eingefärbt, der die Banden im UV-Licht sichtbar macht. Dadurch und durch den Vergleich mit dem Größenmarker kann die Länge und Häufigkeit der erhaltenen DNA Fragmente ermittelt werden. Auswertung der in Abbildung 2 gezeigten Ergebnisse: Die unbehandelten DNA-Proben wurden wie erwartet nicht von einem Restriktionsenzym geschnitten, und weisen alle eine Länge von etwa 260 bp auf. Das Restriktionsenzym BstN1 scheidet in der Erkennungssequenz 5'-CCTGG-3'. Diese Sequenz liegt innerhalb des PLP1-Gens. Bei nicht-mutierten Allelen ist die Erkennungssequenz intakt, und das Enzym kann schneiden. Im mutierten Allel führt die Basensubstitution jedoch dazu, dass die Erkennungssequenz nicht mehr von BstN1 gefunden wird. Mutierte Allele werden also nicht geschnitten. Die Kontroll-DNA trägt keine Mutation auf dem PLP1-Gen in beiden Allelen. Daher wird die DNA-Probe hier an zwei Stellen geschnitten, und es entstehen zwei Fragmente gleicher Länge. Daher lässt sich in der Kontrolle eine dicke Bande mit einer Länge von etwa 130 bp erkennen. Die Mutter besitzt ebenfalls eine Bande mit der Länge von 130 bp, aber auch eine mit der Länge 260 bp. Daher konnte das Restriktionsenzym bei ihr nur in einem Allel schneiden, und sie besitzt ein gesundes und ein mutiertes Allel. Beide Banden sind im Vergleich zu den anderen Proben blass, da hier jeweils nur ein Teil der DNA vorliegt. Der Sohn zeigt nur eine Bande, die sich auf derselben Höhe befindet, wie bei unbehandelten Proben. Das bedeutet, dass der Sohn eine Mutation trägt, und das Restriktionsenzym nicht schneiden konnte. Genotypen von Mutter und Sohn:
- Mutter: Die Mutter weist ein gesundes und ein mutiertes Allel auf und hat somit den Genotyp Xx. Sie ist heterozygot in Bezug auf das mutierte Allel.
- Sohn: Der Sohn besitzt den Genotyp Xy und ist homozygot in Bezug auf das mutierte Allel.
3.
Bedeutung der Myelinscheiden für die Erregungsweiterleitung:
Myelinscheiden sind eine schützende Schicht, die Axone in Wirbeltieren umfasst. Sie schützt das in ihr liegende Axon, und isoliert es elektrisch von der Umgebung. Nur an manchen Stellen – den Ranvierschen Schnürringen – ist diese Schicht unterbrochen. An diesen Schnürringen sind spannungsabhängige Ionenkanäle lokalisiert. Nur an diesen Stellen können Aktionspotenziale ausgelöst werden. Das ermöglicht die schnelle saltatorische Erregungsleitung, bei der das Signal von Schnürring zu Schnürring springt.
Neurophysiologische Auswirkungen der PLP1-Mutation bei Mäusen:
Abbildung 3 zeigt den prozentualen Anteil myelinisierter Axone in Wildtyp- und transgenen Mäusen im Alter von 40, 60 und 120 Tagen. Wildtyp-Mäuse zeigen einen über den gesamten Beobachtungszeitraum recht konstanten Anteil myelinisierter Axone von etwa 96 %. Transgene Mäuse, die eine Mutation des PLP1-Gens erhielten, zeigten schon ab einem Alter von 40 Tagen nur eine Myelinisierungsrate von 60 %. Mäuse in einem Alter von 60 Tagen besaßen nur noch 38 % und 120 Tage alte Mäuse gar keine myelinisierten Axone mehr. Abbildung 4 zeigt die Dauer der Erregungsleitung am Sehnerv in ms von 40 Tage alten Wildtypmäusen im Vergleich zu transgenen Mäusen. An der Abbildung kann abgelesen werden, dass die Dauer der Erregungsleitung von 3 ms bei Wildtypmäusen auf 6 ms bei transgenen Mäusen steigt. Das entspricht einer Verdopplung der Leitungsdauer. Vermutlich wirkt sich die Mutation durch eine drastische Verschlechterung der Sehfähigkeit bis hin zur Erblindung transgener Mäuse aus.
Erklärung der Symptome einer PLP1-Mutation beim Menschen:
Die Funktion des PLP1-Proteins ist in Menschen vermutlich analog zu der in Mäusen, da sich die DNA-Sequenz des Gens nicht unterscheidet. Daher kann auch in Meschen davon ausgegangen werden, dass eine Mutation im PLP1-Gen die Myelinisierungsrate der Axone herabsetzt und Patienten eine deutlich verlangsamte Erregungsleitung zeigen. Fehlt die schützende Myelinscheide um die Axone, sind diese anfälliger für etwaige Beschädigungen und die elektrische Isolierung ist beeinträchtigt. Personen, die an PMK erkrankt sind, zeigen daher Symptome wie eine verzögerte geistigen und körperlichen Entwicklung, sowie Lähmungen der Muskulatur. Das liegt daran, dass das Zentralnervensystem nicht so effizient agieren kann, wie in gesunden Personen, und Signale langsamer oder nur abgeschwächt zwischen Gehirn und Körper ausgetauscht werden.