Vorschlag A – Spinale Muskelatrophie
Spinale Muskelatrophie (SMA)
Die spinale Muskelatrophie (SMA) ist eine vererbbare Krankheit, die zu einem fortschreitenden Abbau der Skelettmuskulatur führt. Die Ursache dieses Muskelabbaus liegt im Absterben der motorischen Nervenzellen, deren Zellkörper sich im Rückenmark („spinal“) befinden. Je nach SMA-Typ kann die Krankheit mild verlaufen oder aber auch tödlich enden. Bis vor Kurzem erfolgte eine Behandlung überwiegend über Physiotherapie, jetzt gibt es neue Ansätze auf Basis der Gentherapie.Genetische Ursachen und Behandlung der Spinalen Muskelatrophie
1
Beschreibe den chemischen und räumlichen Aufbau der DNA.
(8 BE)
2
Beschreibe die wesentlichen Vorgänge der Transkription und der Reifung der mRNA bei Eukaryoten. Erläutere den Zusammenhang zwischen der bei SMA-Patienten vorliegenden Mutation und dem Auftreten von SMA. (Material 1)
(13 BE)
3
Analysiere die molekulargenetischen Ursachen für die Bildung des instabilen SMN-Proteins aus dem SMN2-Gen. (Material 2 und Code-Sonne der mRNA)
(7 BE)
4
Entwickle eine Hypothese, die einen Zusammenhang zwischen der Anzahl der SMN2-Genkopien und dem Auftreten der verschiedenen SMA-Typen herstellt. (Material 1, 2 und 3)
(7 BE)
5
Beschreibe das Prinzip der Gelelektrophorese. Analysiere das Ergebnis der molekulargenetischen Untersuchung von vier Personen im Zusammenhang mit dem Auftreten von SMA und leite daraus den Vererbungsmodus der SMA her. (Material 1, 2 und 4)
(16 BE)
6
Erkläre anhand von Material 5 die Wirkungsweise der beiden gentherapeutischen Behandlungsmethoden von SMA. (Material 1, 2 und 5)
(12 BE)
7
Diskutiere den Einsatz beider Gentherapeutika, auch unter Berücksichtigung von Material 6. (Material 1, 3, 5 und 6)
(8 BE)
Neurophysiologische Ursachen der Spinalen Muskelatrophie
8
Skizziere den Aufbau eines Wirbeltierneurons und nenne die Funktion von sechs Strukturen dieser Zelle.
(9 BE)
9
Werte die in Material 7 und in den Abbildungen 8.1 und 8.2 dargestellten Untersuchungsergebnisse hinsichtlich des Einflusses des SMN-Proteins aus. (Material 7 und 8)
(10 BE)
10
Erkläre die Bedeutung des SMN-Proteins für die normale Muskelfunktion auf molekularbiologischer Ebene. (Material 1, 7 und 8)
(10 BE)
(100 BE)
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Material 1
Genetische Ursachen der Spinalen Muskelatrophie (SMA)
Patientinnen und Patienten, die an SMA leiden, erleben nach und nach den Verlust der Funktion ihrer Skelettmuskulatur. Der Zeitpunkt, ab dem die Krankheit SMA ausbricht, ist unterschiedlich, zumeist beginnt die Krankheit aber schon im Baby- oder Kleinkindalter. Je nach vorher erlernten Bewegungsmustern verlieren die Betroffenen die Fähigkeit zu gehen oder zu sprechen und bei schwerem Verlauf auch zu atmen. Alle Betroffenen können keine ausreichende Menge eines bestimmten Proteins, des SMN-Proteins, bilden. SMN steht für „survival motor neuron“ und gibt damit sehr gut die Bedeutung des Proteins wieder: Ohne dieses Protein sterben motorische Nervenzellen im Rückenmark, dieMaterial 2
Das SMN2-Gen als fast identische Genkopie
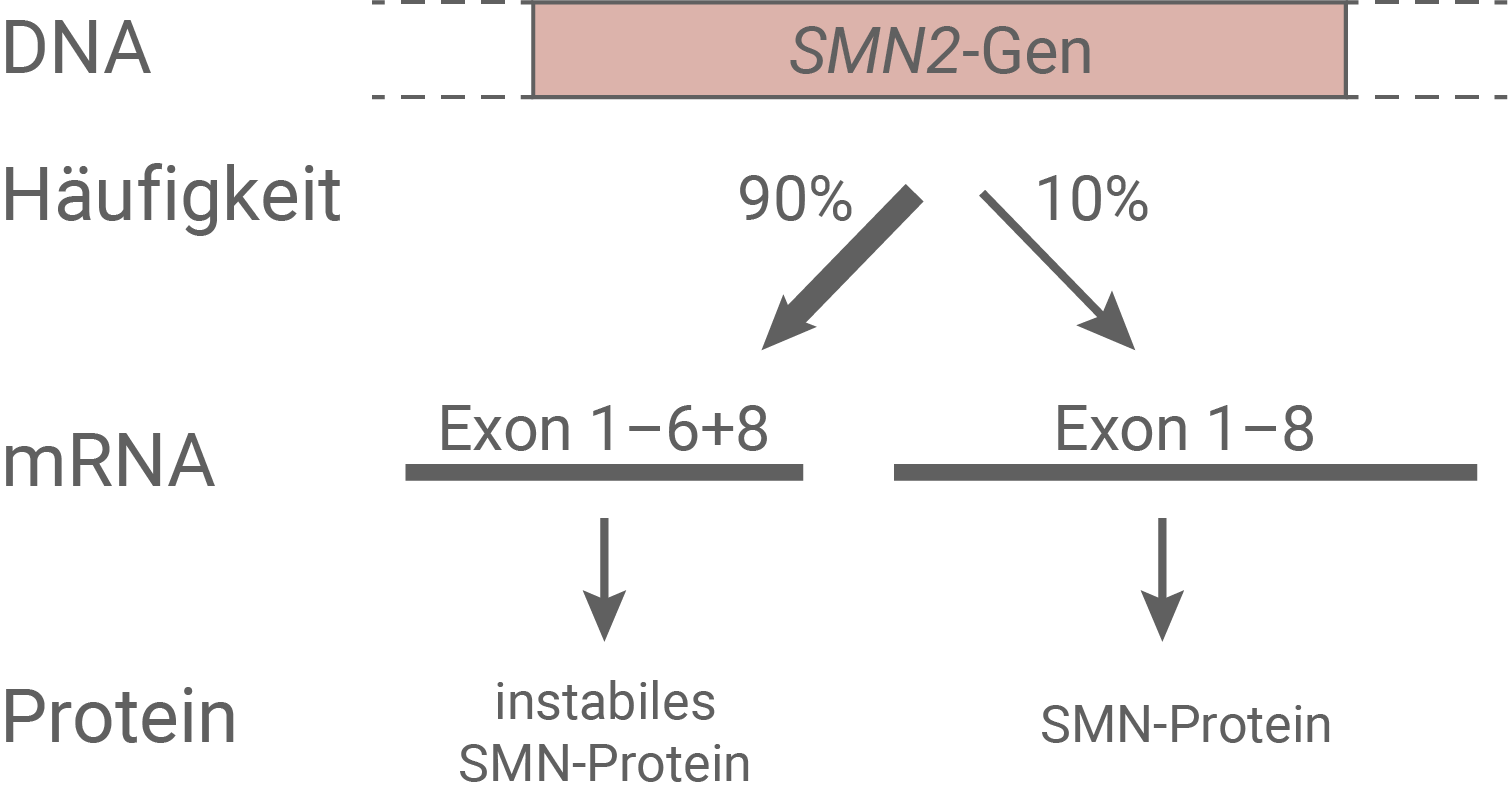
Neben dem SMN1-Gen besitzen alle Menschen eine zweite Variante des SMN-Gens auf Chromosom 5, nämlich das SMN2-Gen. Durch Duplikation kann das SMN2-Gen in unterschiedlicher Anzahl (eins bis sechs) vorkommen. Bei Personen, die eine Deletion von Exon 7 im SMN1-Gen besitzen, ist die Existenz des SMN2-Gens von besonderer Bedeutung. Beide Varianten, das SMN1-Gen und das SMN2-Gen, stimmen zu über 99 % überein. Das vom SMN2-Gen codierte Protein ist jedoch überwiegend instabil und wird rasch abgebaut. Die Ursache dafür liegt in einer Mutation in Exon 7 des SMN2-Gens. Exon 7 umfasst die Nukleotidpositionen 868 bis 921.Die Nukleotid-Sequenzen von Exons enthalten neben den Protein-codierenden Informationen gerade zu Beginn der Exonbereiche auch entscheidende Informationen für die Regulation des Spleißvorgangs im Zellkern.
Abb. 2.1: Ausschnitt aus Exon 7 des codogenen Strangs des SMN1- und des SMN2-Gens
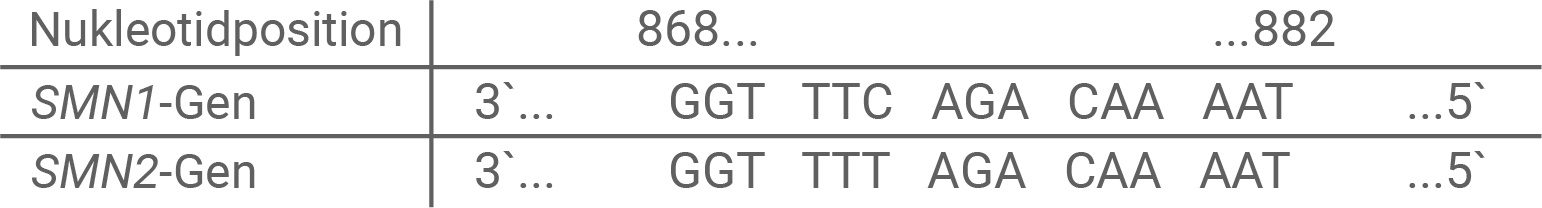
Abb. 2.2: Expression des SMN2-Gens
Material 3
Typen der SMA
In Europa werden jedes Jahr ungefähr 550-600 Säuglinge mit SMA geboren. Der Verlauf der Erkrankung ist dabei sehr variabel. Abhängig vom Beginn der Erkrankung, von den erreichten motorischen Entwicklungsschritten und von der Schwere des Verlaufes werden drei SMA-Typen unterschieden, die in der folgenden Tabelle aufgeführt sind. Unbehandelt führt die schwerste Form bei 90 % der Fälle bis zum Alter von zwei Jahren zum Tod oder zur Notwendigkeit einer ständigen Beatmung. Bei der molekulargenetischen Untersuchung von zahlreichen an SMA erkrankten Personen wurde ein Zusammenhang zwischen der Anzahl der SMN2-Genkopien und dem Schweregrad der Erkrankung festgestellt.Kriterien für die Einteilung der SMA

Material 4
Molekulargenetischer Nachweis der SMA
Mittlerweile ist es möglich, die Krankheit durch einen Gentest eindeutig zu diagnostizieren. Dazu reichen schon wenige Blutstropfen des Neugeborenen. Die daraus gewonnene DNA wird mittels PCR-Methode vervielfältigt und anschließend durch Gelelektrophorese analysiert. Die PCR führt zu Genabschnitten, die das Exon 7 einschließen und dann eine Größe von 200 Basenpaaren aufweisen. Bei Personen, die eine Deletion von Exon 7 SMN1-Gen aufweisen, wird dieser Genabschnitt nicht vervielfältigt. Eine Schwierigkeit im Fall der SMA-Diagnose liegt darin, dass das SMN2-Gen exakt die gleiche Länge wie das intakte SMN1-Gen besitzt. Durch die Mutation im SMN2-Gen ergibt sich jedoch eine weitere Schnittstelle für ein Restriktionsenzym. Bei der molekulargenetischen Untersuchung auf SMA bei vier verschiedenen Personen, die nicht aus einer Familie stammen, konnte durch ein spezielles Verfahren, bei dem auch das oben genannte Restriktionsenzym verwendet wurde, die jeweilige Menge der untersuchten DNA-Fragmente ermittelt werden. Nur Person 3 zeigt phänotypisch die Symptome der SMA.Ergebnis der Gelelektrophorese bei der Untersuchung von vier Personen auf SMA

Material 5
Gentherapie bei SMA
Um ein Fortschreiten der potenziell tödlichen Krankheit aufzuhalten bzw. eine Verbesserung bereits bestehender Symptome zu erzielen, sind in den letzten Jahren Medikamente entwickelt worden, die auf gentherapeutischer Basis wirken. Zwei sowohl in den USA als auch in Europa zugelassene Gentherapeutika basieren auf unterschiedlichen Konzepten. Derzeit gibt es intensive Forschungen dazu, das Grundkonzept beider Methoden auch auf andere genetisch bedingte Erkrankungen zu übertragen.A Gen-Therapie mit Antisense-Oligonukleotiden (ASO)
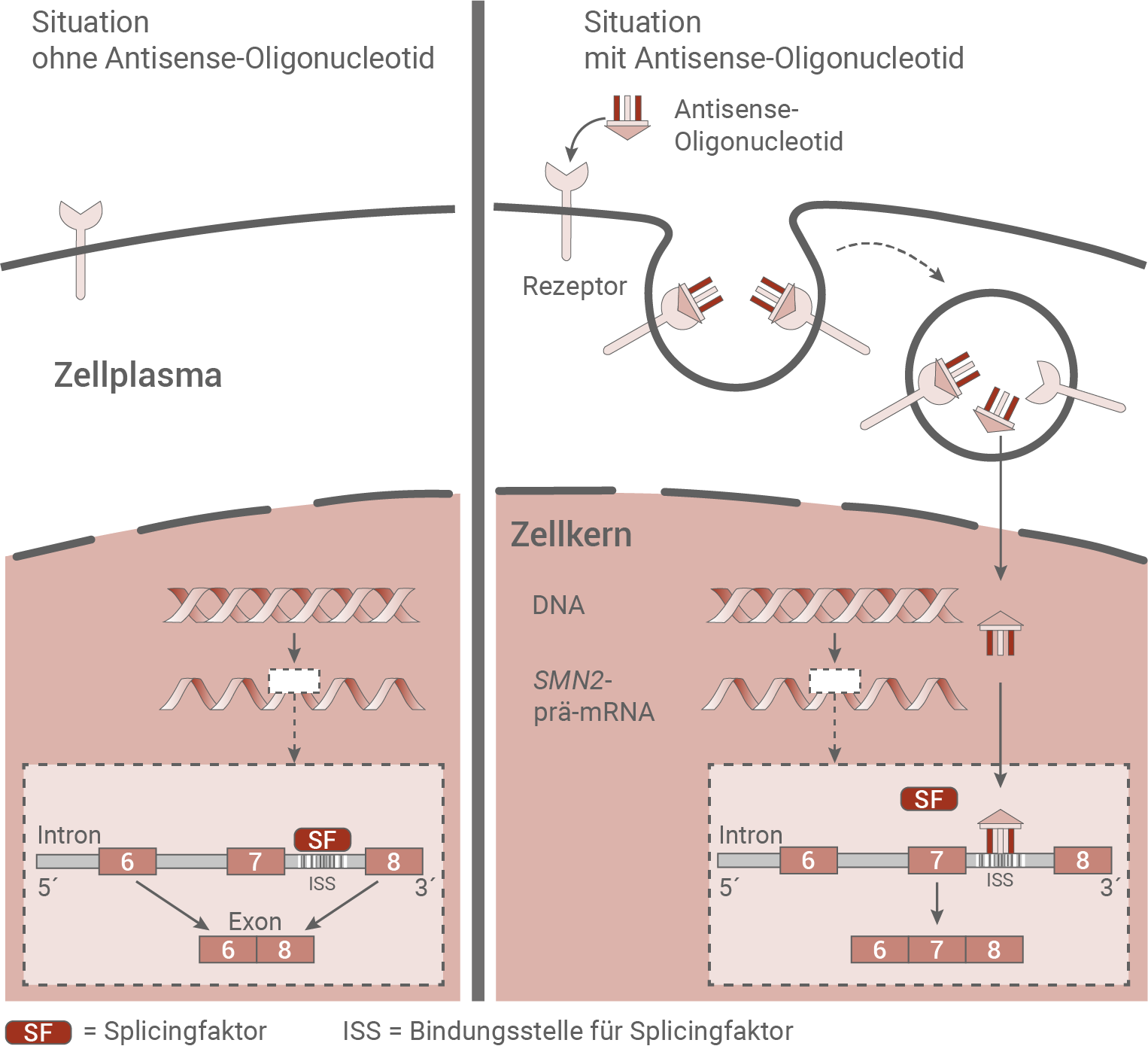
ASO sind künstlich hergestellte, kurze, einzelsträngige DNA- oder RNA-Abschnitte. Der Wirkstoff Nusinersen ist ein ASO mit einer Länge von 18 RNA-Nukleotiden, der an einen Proteinanteil gekoppelt ist. Mehrere Studien beweisen die Wirksamkeit des ASO Nusinersen sowohl bei Kleinkindern als auch bei Erwachsenen. 40 % der Patienten zeigten nach 14-monatiger Behandlung klinisch bedeutsame Verbesserungen ihrer Motorik. Schwerwiegende Nebenwirkungen wurden nicht beobachtet. Die Verabreichung des Wirkstoffs erfolgt durch Injektion direkt in den Rückenmarkskanal zu den Zellkörpern derAbb. 5.1: Wirkmechanismus des ASO Nusinersen
B Gen-Therapie mit Zolgensma 
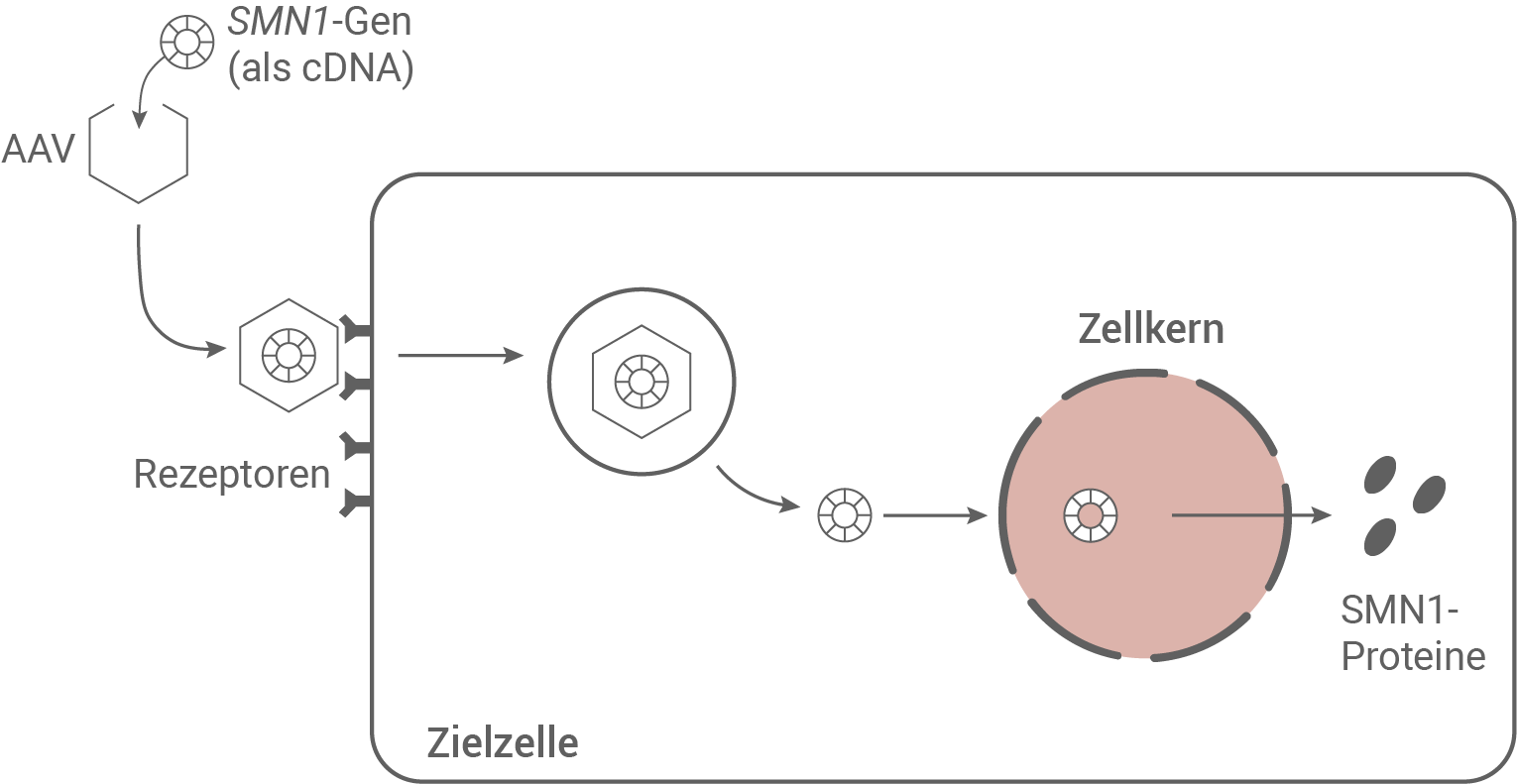
Seit 1. Juli 2020 ist der Wirkstoff Onasemnogen-Abeparvovec (Zolgensma Abb. 5.2: Wirkmechanismus von Zolgensma 
Hinweis
Die cDNA (circuläre DNA) verbleibt dauerhaft im Zellkern und wird nicht in die DNA der Zielzelle integriert.
Die cDNA (circuläre DNA) verbleibt dauerhaft im Zellkern und wird nicht in die DNA der Zielzelle integriert.
Material 6
Zugang aller Menschen zu unverzichtbaren Medikamenten
„Gesundes Leben für alle“ ist eines der Ziele für nachhaltige Entwicklung, die die Vereinten Nationen 2016 formuliert haben. Dieses schließt ein, den Zugang aller Menschen zu unentbehrlichen Arzneimitteln sicherzustellen.Unentbehrliche Arzneimittel sind nach der Definition der Weltgesundheitsorganisation (WHO) solche Arzneimittel, die benötigt werden, um die dringlichsten Bedürfnisse der Bevölkerung zur medizinischen Versorgung zu befriedigen. Sie sollen in einem Gesundheitssystem in adäquater Menge, richtiger Dosierungsform, guter Qualität und zu einem für den Patienten erschwinglichen Preis verfügbar sein. Dazu gehören neben Schmerzmitteln auch Wirkstoffe gegen die häufigsten Infektionskrankheiten, z. B. Antibiotika. Weiterhin sterben jährlich schätzungsweise 1,5 Millionen Kinder unter fünf Jahren weltweit an Krankheiten wie Masern, Diphtherie, Keuchhusten, Kinderlähmung und an weiteren der häufigsten Infektionskrankheiten, vor denen sie durch verfügbare Impfungen geschützt gewesen wären. Eine Dosis eines Kombinations-Impfstoffes, der vor fünf Infektionskrankheiten schützt, kostet beispielsweise weniger als 70 Cent. Dennoch hat gemäß einer Veröffentlichung der Weltbank ein Drittel der Weltbevölkerung keinen effektiven Zugang zu unentbehrlichen Arzneimitteln.
Material 7
Entwicklung von Axonen unter dem Einfluss des SMN-Proteins
Am Institut für Klinische Neurobiologie der Universität Würzburg untersuchte eine Forscherin die Rolle des SMN-Proteins für das Axonwachstum. Um den bei SMA-Patienten vom Typ I vorherrschenden Bedingungen möglichst nahe zu kommen, wurden alle Versuche anAbb. 7.1: Längenwachstum von Axonen
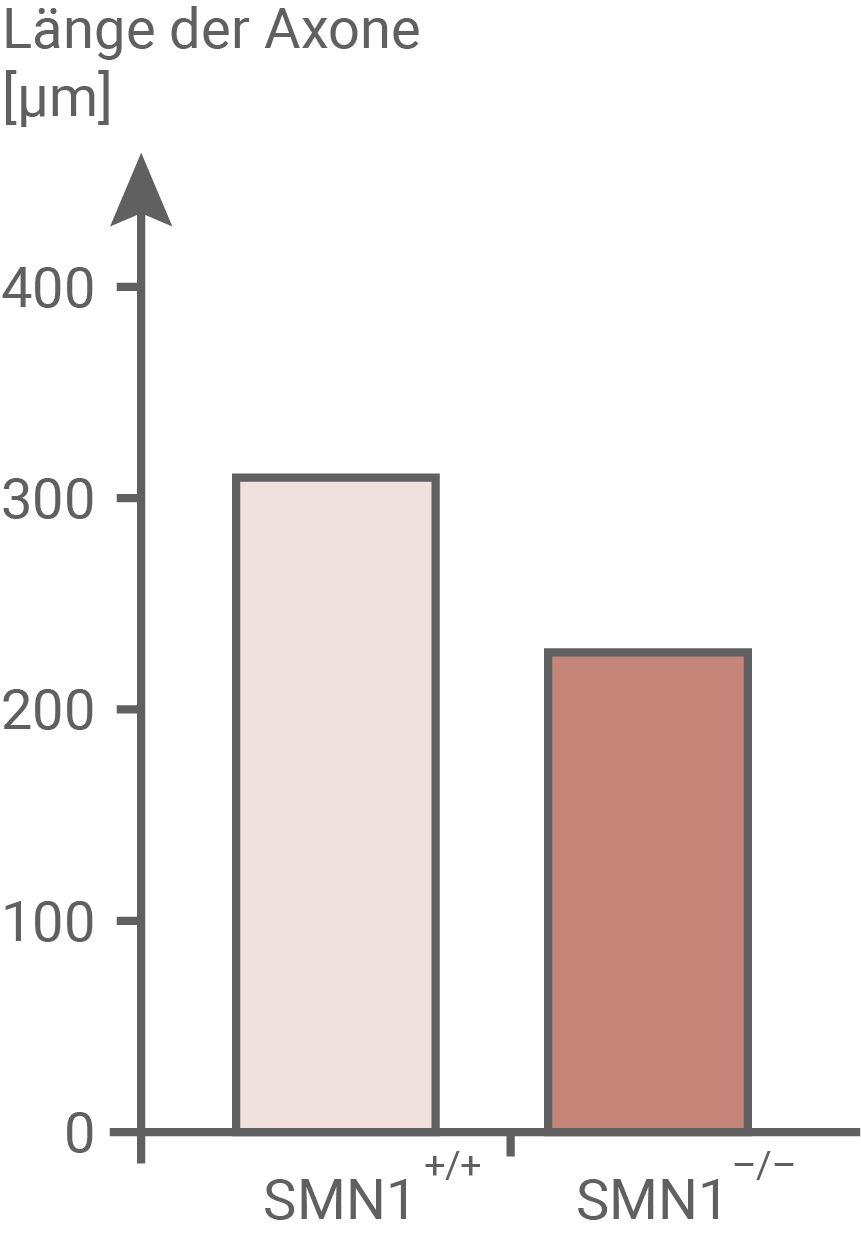
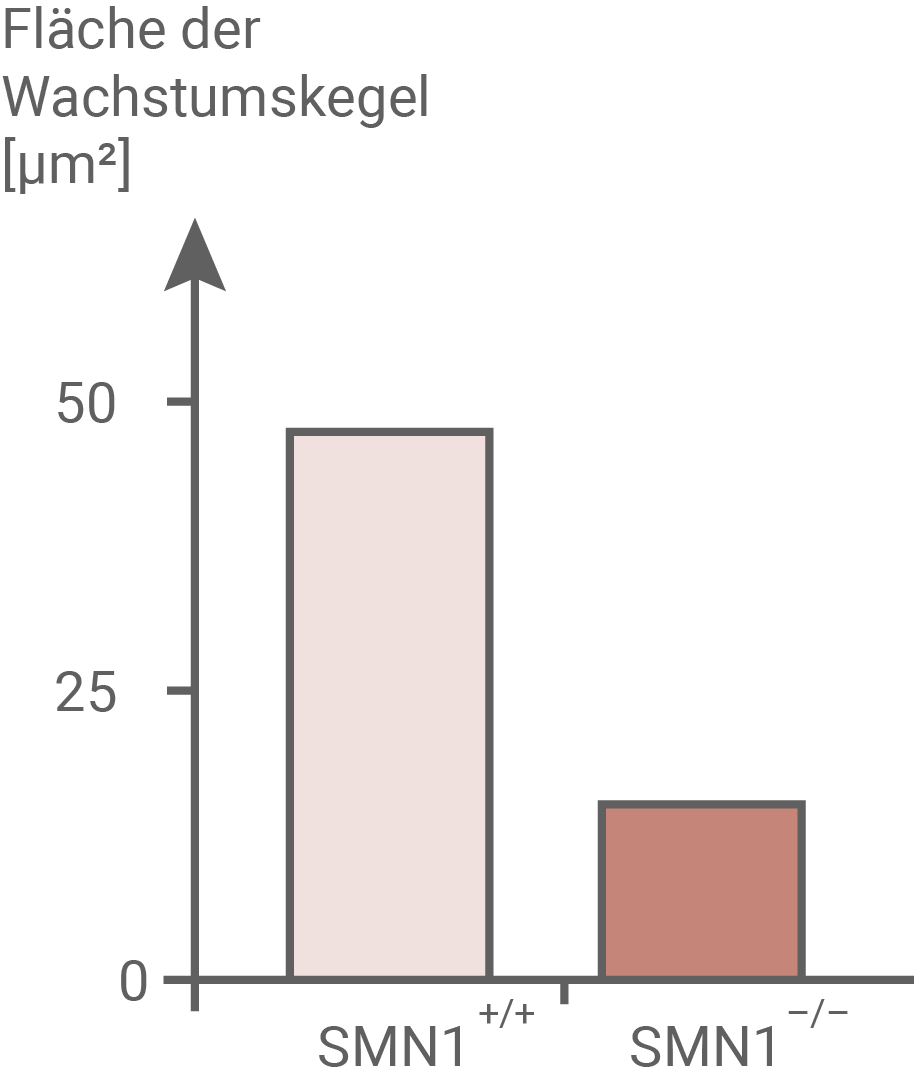
Abb. 7.2: Fläche der Wachstumskegel von Motoneuronen
Wachstumskegel bilden die Endpunkte der Axone, wo zunächst das gerichtete Längenwachstum stattfindet und die später als präsynaptische Bereiche die Verbindung zum Zielgewebe herstellen.
Material 8
Mikrofilamente, Nervenfunktion und SMN-Protein
Im Inneren jeder Zelle befindet sich das Zytoskelett. Es ist ein aus Proteinen aufgebautes Netzwerk im Zytoplasma jeder Zelle, das aus dünnen, fadenförmigen Zellstrukturen (Filamenten) besteht. Eine Klasse dieser Filamente, die Aktinfilamente, sind in Nervenzellen überwiegend aus dem ProteinForschungen haben gezeigt, dass
Das Vorkommen von
Abb. 8.1: Verteilung von  -Aktin-Protein in
-Aktin-Protein in  -Motoneuronen
-Motoneuronen
Ein weiterer Forschungsgegenstand ist die Beantwortung der Frage, wie die im Zellkern einer Nervenzelle gebildete mRNA bis zum Zielort der benötigten Proteine, beispielsweise an der motorischen Endplatte, transportiert wird. Forscher identifizierten in diesem Zusammenhang ein spezielles Transportprotein (RNP), das 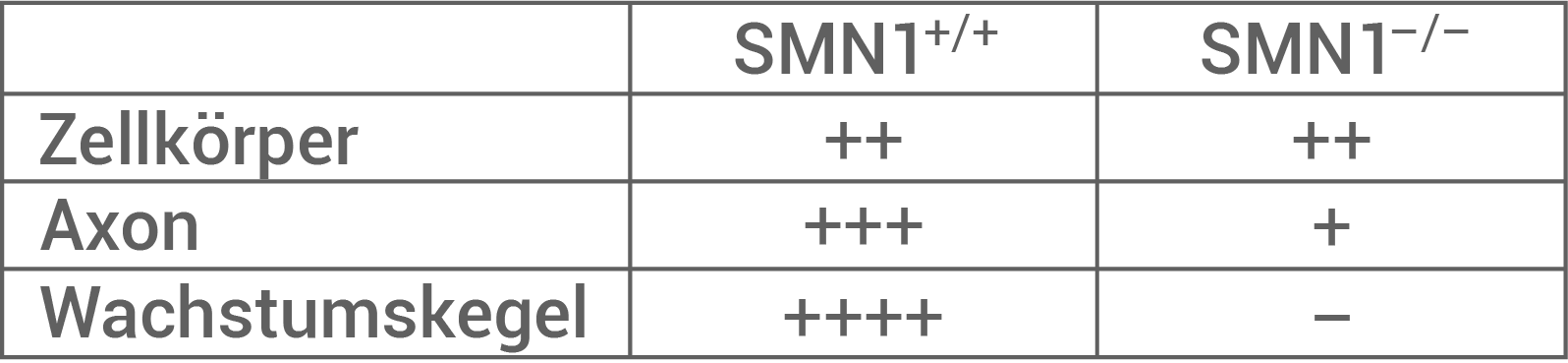
Hinweis
Die Anzahl der + symbolisiert die Menge an Protein, – bedeutet, dass kein Protein vorhanden ist.
Ein weiterer Forschungsgegenstand ist die Beantwortung der Frage, wie die im Zellkern einer Nervenzelle gebildete mRNA bis zum Zielort der benötigten Proteine, beispielsweise an der motorischen Endplatte, transportiert wird. Forscher identifizierten in diesem Zusammenhang ein spezielles Transportprotein (RNP), das Die Anzahl der + symbolisiert die Menge an Protein, – bedeutet, dass kein Protein vorhanden ist.
Abb. 8.2: Verteilung des RNP-Transportproteins in -Motoneuronen
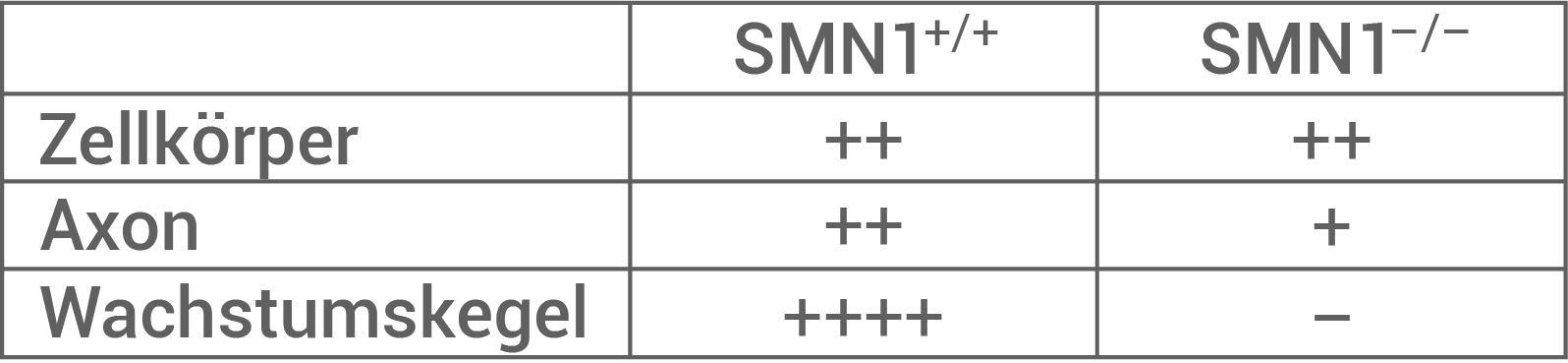
Hinweis
Die Anzahl der + symbolisiert die Menge an Protein, – bedeutet, dass kein Protein vorhanden ist.
Die Anzahl der + symbolisiert die Menge an Protein, – bedeutet, dass kein Protein vorhanden ist.
Abb. 8.3: Funktionsmodell des SMN-RNP-mRNA-Komplexes
Das folgende Funktionsmodell zeigt die mögliche Interaktion der beteiligten Moleküle beim Transport der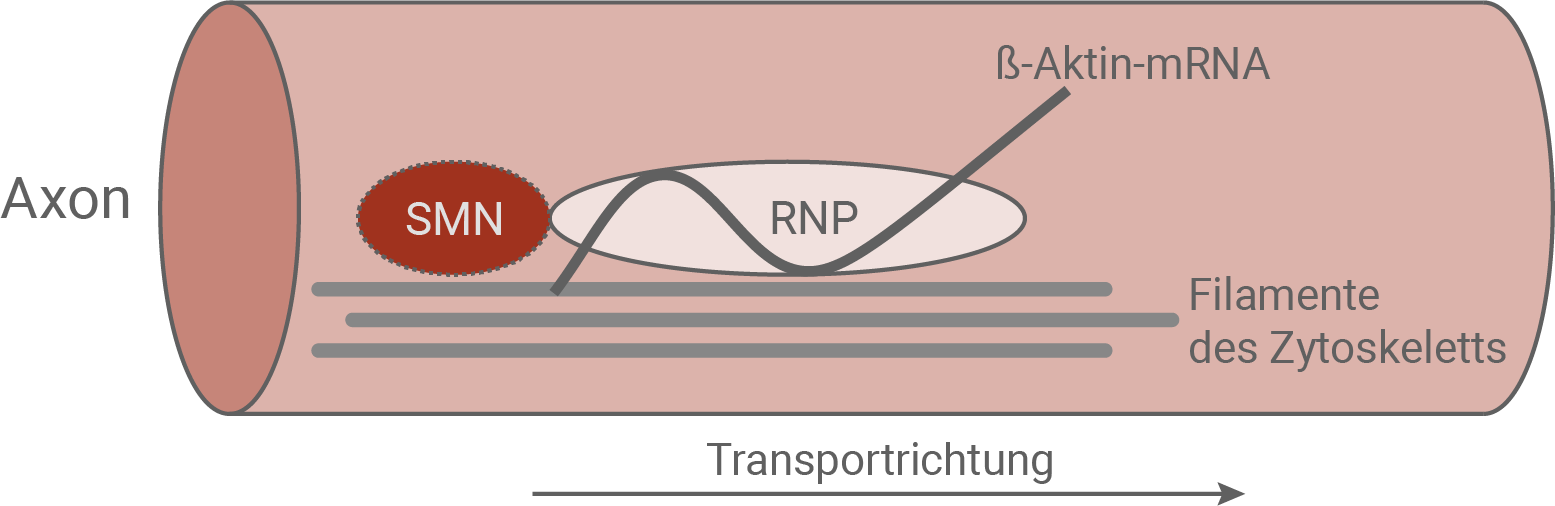
Code-Sonne der mRNA

Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?
1
Chemischer und räumlicher Aufbau der DNA
Die DNA besteht aus Nukleotiden, die sich aus dem Zuckermolekül Desoxyribose, einem Phosphatrest und einer der vier organischen Basen Adenin, Cytosin, Guanin und Thymin zusammensetzen. Das Zuckermolekül Desoxyribose besteht aus insgesamt 5 Kohlenstoffatomen. Nummeriert man diese durch, so ist am C1-Atom stets eine der vier Basen gebunden. Zwischen dem 3. Kohlenstoffatom des ersten, und dem 5. Kohlenstoffatom des darauffolgenden Desoxyribosemoleküls befindet sich der Phosphatrest. In einem Einzelstrang sind die Nukleotide so angeordnet, dass abwechselnd ein Zucker- und Phosphatmolekül nach außen zeigen, und das Rückgrat bilden. Dadurch kann den Einzelsträngen eine Strichrichtung zugeordnet werden, wobei die Einzelstränge antiparallel zueinander verlaufen. Die DNA liegt in Form einer Doppelhelix vor, welche in ihrem Inneren durch Wasserstoffbrücken der Basen verbunden ist. Dabei bindet je eine der Purinbase (Adenin und Guanin) mit einer Pyrimidinbase (Thymin und Cytosin). Adenin und Thymin binden über zwei Wasserstoffbrücken, wohingegen Guanin und Cytosin über drei Wasserstoffbrücken binden.
2
Transkription und Reifung der mRNA bei Eukaryoten
Die Transkription beschreibt die Synthese eines mRNA-Strangs aus einem Teil der DNA, und lässt sich in drei Abschnitte einteilen.
- Initiation: Die RNA-Polymerase öffnet durch Auflösen der Wasserstoffbrücken den DNA-Doppelstrang an der Promotorregion. Die Polymerase beginnt mit der Synthese des mRNA-Strangs am Startpunkt auf dem codogenen Strang.
- Elongation: Die im Zellkern vorhandenen RNA-Nukleotide lagern sich an den codogenen Strang an. Sie unterscheiden sich insofern von DNA-Nukleotiden, als dass hier Ribose als Zuckermolekül, und die Base Uracil anstelle von Thymin verwendet wird. Die Nukleotide sind zum codogenen Strang komplementär. Die DNA-Polymerase verknüpft die Nukleotide in 5'
3'-Richtung, wobei sich das Enzym selbst in 3'
- Termination: Trifft die RNA-Polymerase auf die Terminatorregion, so endet die Synthese des mRNA-Stranges. Die RNA-Polymerase löst sich von der DNA, und die prä-mRNA wird freigesetzt.
3
Molekulargenetische Ursachen für die Bildung des instabilen SMN-Proteins aus dem SMN2-Gen:
Im Vergleich der Nukleotidsequenzen des SMN1- und des SMN2-Gens, lässt sich eine Mutation der 3. Position im zweiten Triplett der Nukleotidsequenz des SMN2-Gens feststellen. Statt Cytosin wurde die Base Guanin eingebaut, es handelt sich somit um eine Punktmutation. Die Mutation prägt sich folgendermaßen bei der Transkription und der Translation aus:
- SMN1-Gen:
mRNA: CCA AAG UCU GUU UUA
ASS: Pro Lys Ser Val Leu - SMN2-Gen:
mRNA: CCA AAA UCU GUU UUA
ASS: Pro Lys Ser Val Leu
4
Hypothese zum Zusammenhang zwischen der Anzahl der SMN2-Genkopien und dem Auftreten der SMA-Typen
Spinale Muskelatrophie (SMA) wird durch die Deletion im SMN1-Gen und das infolgedessen funktionsunfähige SMN-Protein hervorgerufen. Durch die Expression des SMN2-Gens kann dennoch eine gewisse Menge des benötigten SMN-Proteins hergestellt werden. Dadurch kann das Fehlen des durch das SMN1-Gen exprimierten, funktionsunfähigen SMN-Gens zum Teil kompensiert werden. Je mehr Genkopien es von dem SMN2-Gen gibt, desto höher ist die Wahrscheinlichkeit, dass ausreichend SMN-Protein gebildet wird. Je mehr Protein gebildet wird, desto schwächer sind die Symptome von SMA ausgeprägt, und die motorischen Fähigkeiten sind weniger stark beeinträchtigt. Es besteht somit ein Zusammenhang zwischen der Anzahl der Genkopien des SMN2-Gens und dem Schweregrad der Krankheit.
Beim Typ 1 bricht die Krankheit bereits im Alter von unter 6 Monaten aus. Betroffene Kinder können niemals frei sitzen, da die Skelettmuskulatur zu schwach ist. Der Tod tritt dabei schon im Alter von unter 2 Jahren ein. Vermutlich liegen hier nur sehr wenige Kopien des SMN2-Gens vor, sodass das fehlende Protein nicht kompensiert werden kann.
Beim Typ 2 beginnt die Krankheit in den ersten 18 Lebensmonaten. Betroffene können frei sitzen, werden jedoch niemals frei gehen können. Die Lebenserwartung geht hier über das Kleinkindalter hinaus. Vermutlich haben Betroffene des Typs 2 mehr Genkopien als Betroffene von Typ 1, durch die eine größere Menge des fehlerhaften Proteins kompensiert werden kann.
Beim Typ 3 zeigt sich die Krankheit erst nach 18 Monaten. Betroffenen Personen ist es möglich, frei gehen und stehen zu können, und sie werden das Erwachsenenalter erreichen. Es liegt nahe, dass diese Personen eine ausreichend hohe Anzahl an Genkopien des SMN2-Gens besitzen, um recht uneingeschränkt leben zu können.
Beim Typ 1 bricht die Krankheit bereits im Alter von unter 6 Monaten aus. Betroffene Kinder können niemals frei sitzen, da die Skelettmuskulatur zu schwach ist. Der Tod tritt dabei schon im Alter von unter 2 Jahren ein. Vermutlich liegen hier nur sehr wenige Kopien des SMN2-Gens vor, sodass das fehlende Protein nicht kompensiert werden kann.
Beim Typ 2 beginnt die Krankheit in den ersten 18 Lebensmonaten. Betroffene können frei sitzen, werden jedoch niemals frei gehen können. Die Lebenserwartung geht hier über das Kleinkindalter hinaus. Vermutlich haben Betroffene des Typs 2 mehr Genkopien als Betroffene von Typ 1, durch die eine größere Menge des fehlerhaften Proteins kompensiert werden kann.
Beim Typ 3 zeigt sich die Krankheit erst nach 18 Monaten. Betroffenen Personen ist es möglich, frei gehen und stehen zu können, und sie werden das Erwachsenenalter erreichen. Es liegt nahe, dass diese Personen eine ausreichend hohe Anzahl an Genkopien des SMN2-Gens besitzen, um recht uneingeschränkt leben zu können.
5
Prinzip der Gelelektrophorese:
Das Verfahren der Gelelektrophorese dient der Unterscheidung geladener Teilchen (wie zum Beispiel DNA-Fragmente) nach ihrer Größe. Durch eine anschließende Färbung des Gels können diese Fragmente sichtbar gemacht werden. Die Gelelektrophorese besteht aus einer Kammer, in der sich ein Agarose-Gel befindet. Das Gel ist strukturell wie ein engmaschiges Netz aufgebaut, wodurch größere Fragmente langsamer wandern als kleinere. Auf der Kathodenseite des Gels werden die zu untersuchenden Proben gemeinsam mit einem Ladepuffer in dafür vorgesehene Taschen im Gel gegeben. In der Regel wird in eine äußere Tasche ein Größenmarker gegeben, um die DNA-Fragmente später zuordnen zu können. Im Anschluss wird eine elektrische Spannung angelegt, und die Fragmente beginnen zu wandern. Da die DNA polar und durch ihr Phosphatrückgrat negativ geladen ist, wandert sie im Gel von der Kathode in Richtung Anode. Nach einer definierten Zeitspanne, wird das Gel von der Spannungsquelle getrennt, und mit einem Farbstoff eingefärbt, der die Banden im UV-Licht sichtbar macht. Dadurch und durch den Vergleich mit dem Größenmarker kann die Länge und Häufigkeit der erhaltenen DNA Fragmente ermittelt werden.
Analyse des Ergebnisses der molekularen Untersuchung:
Das SMN1-Gen, welches Exon 7 beinhaltet, ist 200 bp lang, und weist keine Restriktionsschnittstelle auf. Das SMN2-Gen hat ebenfalls eine Länge von 200 bp, durch die Punktmutation ergibt sich hier allerdings eine Restriktionsschnittstelle, wodurch Fragmente mit einer Länge von 176 bp und 24 bp entstehen. Je stärker eine Bande in der Gelelektrophorese ist, desto größer ist die Menge des entsprechenden DNA-Fragments.
Person 1 hat kein intaktes SMN1-Gen. Die Kopien des SMN2-Gens reichen allerdings nicht aus, um die fehlende Proteinmenge auszugleichen. Person 3 ist von SMA betroffen. Herleitung des Vererbungsmodus bei SMA: Aus Material 1 geht hervor, dass die Krankheit autosomal vererbt wird, da der Gendefekt auf Chromosom 5 – einem Autosom – liegt. Aus der Gelelektrophorese geht hervor, dass Personen, die nur über eine einfache Kopie des SMN1-Gens verfügen, und damit heterozygot sind, nicht von der Krankheit betroffen sind, da sie genügend Protein herstellen können. Demzurfolge muss eine Person homozygot bezüglich des nicht vorhandenen SMN1-Gen sein, damit sie von SMA betroffen ist. Die Krankheit wird also autosomal-rezessiv vererbt.
- Bei Person 3 zeigt sich keine Bande bei 200 bp. Daher ist davon auszugehen, dass diese Person kein intaktes SMN1-Gen besitzt.
- Bei Person 2 lässt sich eine dünne Bande bei 200 bp erkennen. Diese Person besitzt daher nur eine einzelne Kopie des SMN1-Gens.
- Die Personen 1 und 4 weisen eine in etwa doppelt so dicke Bande bei 200 bp auf, wie Person 2. Daher besitzen Person 1 und 4 zwei Genkopien des SMN1-Gens.
- Die Personen 1, 2 und 3 haben dickere Banden bei der Länge 176 bp und 24 bp. Daher weisen sie das SMN2-Gen in doppelter Anzahl auf.
- Bei Person 4 sind bei 176 bp und 24 bp noch dickere Banden entstanden. Vermutlich besitzt diese Person das SMN2-Gen also in mehr als zweifacher Kopie.
Person 1 hat kein intaktes SMN1-Gen. Die Kopien des SMN2-Gens reichen allerdings nicht aus, um die fehlende Proteinmenge auszugleichen. Person 3 ist von SMA betroffen. Herleitung des Vererbungsmodus bei SMA: Aus Material 1 geht hervor, dass die Krankheit autosomal vererbt wird, da der Gendefekt auf Chromosom 5 – einem Autosom – liegt. Aus der Gelelektrophorese geht hervor, dass Personen, die nur über eine einfache Kopie des SMN1-Gens verfügen, und damit heterozygot sind, nicht von der Krankheit betroffen sind, da sie genügend Protein herstellen können. Demzurfolge muss eine Person homozygot bezüglich des nicht vorhandenen SMN1-Gen sein, damit sie von SMA betroffen ist. Die Krankheit wird also autosomal-rezessiv vererbt.
6
Wirkungsweise der gentechnischen Behandlungsmethoden von SMA:
1.
Gen-Therapie mit Antisense-Oligonukleotiden (ASO):
Bei dieser Therapie wird der Wirkstoff Nusinersen verwendet. Er besteht aus einer Proteinkomponente und einem ASO mit einer Länge von 18 RNA-Nukleotiden. Der Komplex hat aufgrund seiner RNA-Struktur die Möglichkeit, komplementär an eine weitere RNA zu binden. Wird kein Wirkstoff verabreicht, so kann ein Splicingfaktor direkt an seine Bindestelle andocken. Während der Prozessierung wird Exon 7 aus der prä-mRNA herausgeschnitten. Bei einer Behandlung mit dem Wirkstoff Nusinersen bindet das Antisense-Oligonukleotid an einen Rezeptor an der Zellmembran. Der Rezeptor wird mit dem ASO durch Vesikelbildung ins Zellplasma transportiert, wo das ASO vom Rezeptor abgespalten, und in den Zellkern transportiert wird. Bei der Prozessierung der prä-mRNA blockiert das ASO die Bindestelle für den Splicingfaktor. Die prä-mRNA wird dadurch so gespleißt, dass Exon 7 in der mRNA verbleibt. In der Translation wird dadurch ein funktionsfähiges SMN-Protein synthetisiert.
Bei dieser Therapie wird der Wirkstoff Nusinersen verwendet. Er besteht aus einer Proteinkomponente und einem ASO mit einer Länge von 18 RNA-Nukleotiden. Der Komplex hat aufgrund seiner RNA-Struktur die Möglichkeit, komplementär an eine weitere RNA zu binden. Wird kein Wirkstoff verabreicht, so kann ein Splicingfaktor direkt an seine Bindestelle andocken. Während der Prozessierung wird Exon 7 aus der prä-mRNA herausgeschnitten. Bei einer Behandlung mit dem Wirkstoff Nusinersen bindet das Antisense-Oligonukleotid an einen Rezeptor an der Zellmembran. Der Rezeptor wird mit dem ASO durch Vesikelbildung ins Zellplasma transportiert, wo das ASO vom Rezeptor abgespalten, und in den Zellkern transportiert wird. Bei der Prozessierung der prä-mRNA blockiert das ASO die Bindestelle für den Splicingfaktor. Die prä-mRNA wird dadurch so gespleißt, dass Exon 7 in der mRNA verbleibt. In der Translation wird dadurch ein funktionsfähiges SMN-Protein synthetisiert.
2.
Gen-Therapie mit Zolgensma®:
Der Wirkstoff Zolgensma® besteht im Grunde aus einem intakten SMN1-Gen in Form einer einzelsträngigen cDNA, welche in das Virus AAV, welches als Vektor dient, eingeschleust ist. Das Virus selbst ist nicht vermehrungsfähig und dient lediglich dem Transport der cDNA in die Zielzelle. Dazu dockt es an Rezeptoren in der Membran der Zielzellen an. Daraufhin wird die Membran an dieser Stelle eingestülpt, und das Virus gelangt durch Endocytose in das Zellinnere. Im Zellplasma wird die cDNA aus dem Virus freigesetzt, und gelangt durch die Kernporen direkt in den Zellkern. Hier liegt die cDNA als freies Molekül vor, welches zu einer mRNA transkribiert und im Anschluss an den Ribosomen transliert werden kann. So kann ein funktionsfähiges Protein synthetisiert werden.
Der Wirkstoff Zolgensma® besteht im Grunde aus einem intakten SMN1-Gen in Form einer einzelsträngigen cDNA, welche in das Virus AAV, welches als Vektor dient, eingeschleust ist. Das Virus selbst ist nicht vermehrungsfähig und dient lediglich dem Transport der cDNA in die Zielzelle. Dazu dockt es an Rezeptoren in der Membran der Zielzellen an. Daraufhin wird die Membran an dieser Stelle eingestülpt, und das Virus gelangt durch Endocytose in das Zellinnere. Im Zellplasma wird die cDNA aus dem Virus freigesetzt, und gelangt durch die Kernporen direkt in den Zellkern. Hier liegt die cDNA als freies Molekül vor, welches zu einer mRNA transkribiert und im Anschluss an den Ribosomen transliert werden kann. So kann ein funktionsfähiges Protein synthetisiert werden.
7
Diskussion des Einsatzes der beiden gentherapeutischen Behandlungsmethoden:
Argumente gegen den Einsatz der Medikamente:
- Beide Therapiemethoden sind mit immens hohen Kosten verbunden, Nusinersen verursacht lebenslänglich Kosten von 300.000 € jährlich, und Zolgensma® ist mit 2 Millionen € für eine einmalige Dosis das teuerste Medikament der Welt.
- Die Finanzierung dieses Medikaments ist nur in gut entwickelten Ländern mit funktionierendem Gesundheitssystem und ausreichenden finanziellen Ressourcen möglich. Nur reiche Menschen können sich eine Behandlung leisten.
- In die Entwicklung und Forschung an Behandlungsmethoden für SMA wird sehr viel Geld investiert. Demgegenüber steht die Tatsache, dass diese Erbkrankheit extrem selten ist und die Forschungsgelder auch anderweitig verwendet werden könnten.
- Das Geld könnte beispielsweise in die Finanzierung unentbehrlicher Medikamente gesteckt werden, zu denen Menschen aus bestimmten Ländern keinen Zugang haben, die aber viel mehr Personen helfen könnten. Eine Impfung mit einem Kombinationsimpfstoff kostet zum Beispiel nur 70 Cent pro Dosis.
- Dies würde auch den Zielen der WHO und der Vereinten Nationen entsprechen, dass alle Menschen Zugang zu lebenswichtigen Arzneimitteln haben sollten.
- Dadurch könnte der Tod von 1,5 Millionen Kindern, die jährlich an vermeidbaren Infektionskrankheiten sterben, verhindert werden.
- Betroffenen von SMA kann nur durch diese beiden sehr teuren Medikamente geholfen werden. Es existieren keine Alternativen zu diesen Gentherapien.
- Bei der Gabe von Nusinersen kann bei betroffenen Personen eine signifikante Verbesserung der motorischen Fähigkeiten festgestellt werden, ohne dass Patienten an Nebenwirkungen leiden. Die Behandlung mit Zolgensma® reicht nach einmaliger Verabreichung aus, um die betroffene Person zu heilen. Dadurch können auch Kosten eingespart werden, die zum Beispiel durch lebenslange künstliche Beatmung entstehen.
- Grundsätzlich sollte keine Person von der für sie besten Behandlung ausgeschlossen werden. Das gilt auch für Betroffene von SMA. Auch wenn die Behandlungskosten für diese Patienten höher sind, sollten sich durch den Solidaritätsausgleich der Krankenkassen gedeckt werden.
- Das Geld, welches in die Forschung und Entwicklung für Therapien investiert wird, fällt nur einmalig an. Gleichzeitig lassen sich die entdeckten Prinzipien auf verschiedene Krankheiten übertragen. Die Investition ist also auch mit der Möglichkeit verbunden, mehr schwere und vermeintlich unheilbare Krankheiten zu behandeln. So könnten noch mehr Menschen von dem Invest profitieren.
8
Aufbau eines Wirbeltierneurons:
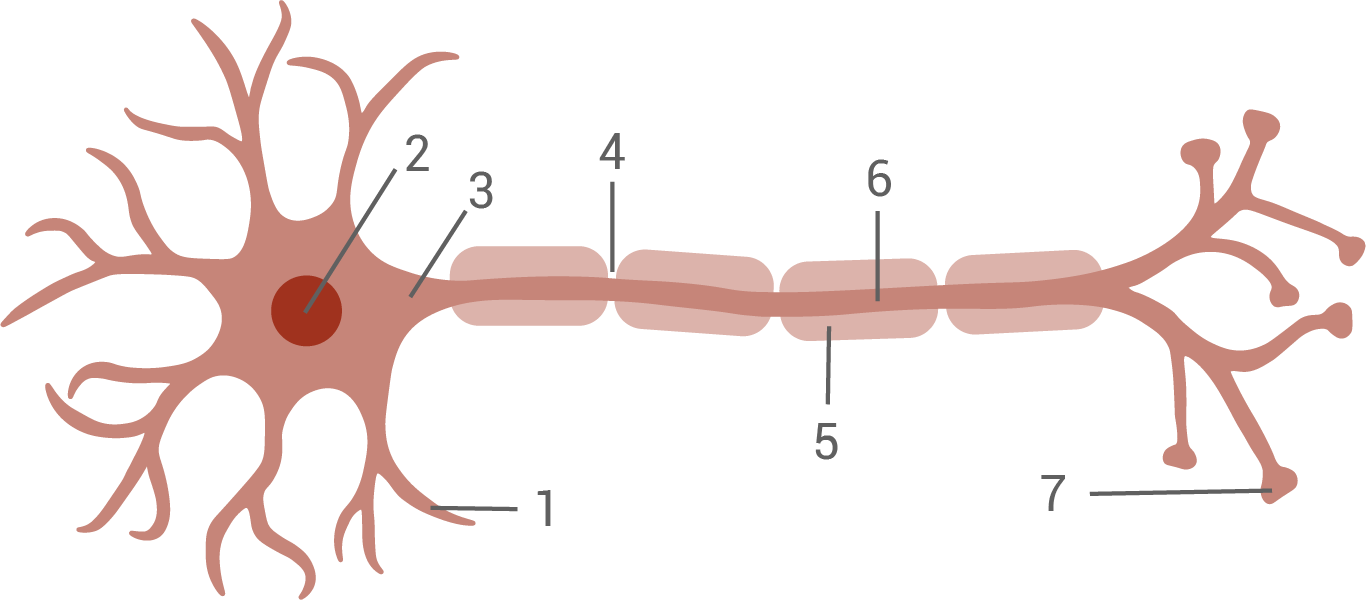
- Dendriten: Sie nehmen Signale von benachbarten Nervenzellen auf, und leiten die Erregung über das Soma an den Axonhügel weiter.
- Zellkern: Der Zellkern ist für die Steuerung aller zentralen Stoffwechselvorgänge zuständig.
- Axonhügel: Hier werden die ankommenden Reize aufsummiert, und gegebenenfalls neue Aktionspotenziale gebildet.
- Ranvierscher Schnürring: Lücken in der Myelinscheide, hier erfolgt die Bildung von Aktionspotenzialen
- Myelinscheide/ Schwannsche Zellen: Dienen zur elektrischen Isolierung des Axons zur schnelleren Erregungsweiterleitung.
- Axon: Weiterleitung der Aktionspotenziale.
- Synaptische Endknöpfchen: Übertragung der vom Axon ankommenden Erregungen an weitere Nervenzellen oder an Muskelfasern über Ausschüttung eines Transmitterstoffs an der Synapse.
Hinweis: Es genügt die Beschriftung und Beschreibung von sechs Strukturen.
9
Auswertung der Untersuchungsergebnisse hinsichtlich des Einflusses des SMN-Proteins:
Daraus kann man schließen, dass sich das SMN-Protein positiv auf das Längenwachstum von Axonen auswirkt.
Das SMN-Protein hat also auch einen erheblichen Einfluss auf die Fläche der Wachstumskegel von  -Motorneuronen hat.
Das SMN-Protein hat demzufolge einen förderlichen Einfluss auf die Bildung von
-Motorneuronen hat.
Das SMN-Protein hat demzufolge einen förderlichen Einfluss auf die Bildung von  -Aktin-Protein im Axon und Wachstumskegel.
Es lässt sich sagen, dass das SMN-Protein einen positiven Einfluss auf die Menge des RNP-Transportproteins in Axon und Wachstumskegel hat.
-Aktin-Protein im Axon und Wachstumskegel.
Es lässt sich sagen, dass das SMN-Protein einen positiven Einfluss auf die Menge des RNP-Transportproteins in Axon und Wachstumskegel hat.
- Aus Abbildung 7.1 geht hervor, dass Individuen mit unverändertem SMN1-Gen Axone mit einer durchschnittlichen Länge von 300
besitze. Genetisch veränderte Mäuse, bei denen das SMN1-Gen homozygot auschgeschaltet wurde, wiesen allerdings, mit nur etwa 220
- Abbildung 7.2 kann man entnehmen, dass Mäuse mit intaktem SMN1-Gen mit etwa 50
einen flächenmäßig mehr als doppelt so großen Wachstumskegel ausbilden als Mäuse mit inaktiviertem SMN1-Gen mit 20
- Abbildung 8.1 zeigt die Menge des vorhandenen
- Abbildung 8.2 zeigt die Verteilung des RNP-Transportproteins im Zellkörper, im Axon und im Wachstumskegel von
10
Bedeutung des SMN-Proteins für die normale Muskelfunktion auf molekularbiologischer Ebene:
Betroffene von SMA leiden unter starker Bewegungseinschränkung, und sind in ihrer motorischen Entwicklung oft eingeschränkt. Bei den Erkrankten ist die Produktion des SMN-Proteins gestört, wodurch -Motorneurone durch Unterversorgung absterben. Das Überleben der -Motorneuronen ist von dem Kontakt mit Muskelfasern abhängig, die die Neurone mit neurotrophen Substanzen versorgen, und somit ein Absterben verhindern. Die Muskelfunktion kann also nur durch intakte -Motorneuronen gewährleistet werden.
Um den Kontakt zu Muskelzellen ausbilden zu können, müssen -Motorneuronen einen Wachstumskegel am Axonende bilden. Für dieses Wachstum sind die dünnen -Aktinfilamente des Cytoskelettes verantwortlich, die sowohl für ein Längenwachstum der Axone und deren Verzweigung als auch für die Ausbildung und Erhaltung von synaptischen Kontakten zuständig sind. Nur bei intakten Synapsen kann eine Erregungsweiterleitung an den Muskel mittels Neurotransmittern erfolgen, und der Muskel bewegt werden.
Die Aufgabe des SMN-Proteins besteht darin, sich an das RNP-Transporterprotein anzulagern. Dieser Komplex ist in der Lage, entlang des Cytoskelettes in Richtung Wachstumskegel zu wandern, und die an das RNP-Transportprotein assoziierte mRNA an den Wachstumskegel zu transportieren. Mithilfe der mRNA kann das -Aktin-Protein hergestellt werden, und steht somit als Filament des Cytoskelettes bereit, welches wiederum beim Wachstum der des Axons Richtung Zielzelle bereitsteht. Durch den entstandenen Kontakt zur Muskelzelle ist die Erregungsübertragung vom synaptischen Endknöpfchen auf die motorische Endplatte gewährleistet. Bei gesunden Personen wird so die Muskelbewegung sichergestellt.