Vorschlag B – Fragiles-X-Syndrom
Fragiles-X-Syndrom
Das Fragile-X-Syndrom, welches eine der häufigsten Ursachen für eine genetisch bedingte Intelligenzminderung ist, wurde 1943 erstmalig von den Ärzten J. P. Martin und J. Bell beschrieben. Als Hauptsymptom gilt eine kognitive Beeinträchtigung, die sehr variabel ist und von einer minimalen Ausprägung bis hin zu einem niedrigen Intelligenzquotienten (IQ) mit ausgeprägter Entwicklungsverzögerung reicht.Genetische Ursachen des Fragilen-X-Syndroms
1
Beschreibe die wesentlichen Vorgänge der Transkription und der Reifung der mRNA bei Eukaryoten sowie den Einfluss der DNA-Methylierung auf die Proteinbiosynthese.
(10 BE)
2
Bestimme alle möglichen Genotypen der Familienmitglieder 1, 2, 5, 6, 11, 17 und 18 im Stammbaum in Material 2 unter Verwendung der in Abbildung 2.1 angegebenen Allelsymbole. Analysiere anhand des Stammbaums die Entwicklung des Gendefekts über mehrere Generationen hinweg unter Einbezug von Abbildung 2.2 und entwickle eine Hypothese zur Ursache der Verlängerung der Repeats. (Material 1 und 2)
(13 BE)
3
Beschreibe das Prinzip der Gelelektrophorese. Ordne die Bandenmuster des molekulargenetischen Nachweises in Abbildung 2.3 exemplarisch jeweils einer Person des Stammbaums (Abbildung 2.1) begründet zu. (Material 2)
(13 BE)
4
Gib die Aminosäuresequenzen des FMR1-Protein-Ausschnitts einer gesunden Person und des Patienten mit Fragilem-X-Syndrom an. (Material 3)
(4 BE)
5
Erläutere für beide Formen des Fragilen-X-Syndroms die Auswirkungen der jeweiligen Mutation auf Bildung und Struktur des FMR1-Proteins (FMRP) und leite daraus einen konkreten Zusammenhang zu der Entstehung des Krankheitsbilds her. (Material 1, 2 und 3)
(11 BE)
Neurophysiologische Ursachen des Fragilen-X-Syndroms
6
Beschreibe zum einen die Erregungsübertragung an einer erregenden, Acetylcholin führenden Synapse und zum anderen die Vorgänge an der postsynaptischen Membran, die zur Entstehung eines IPSP führen.
(11 BE)
7
Erläutere die Funktionsweise der in Material 4 dargestellten Glutamat führenden Synapse sowie die Folgen der beiden Mutationsformen im FMR1-Gen für die Funktion dieser Synapse. Deute die bei Fragilem-X-Syndrom auftretenden Symptome unter neurophysiologischen Gesichtspunkten. (Material 3, 4 und 5)
(17 BE)
8
Leite eine mögliche Wirkungsweise für ein im synaptischen Spalt wirkendes Medikament her, welches die Symptome der Betroffenen mildern könnte. (Material 4)
(4 BE)
9
Fasse die Untersuchungsergebnisse der Studien zur neuronalen Morphologie (Abbildung 7.3 und 7.4) zusammen und analysiere das Untersuchungsergebnis in Abbildung 7.5. Deute die Ergebnisse hinsichtlich der Gedächtnisleistung. (Material 6 und 7)
(17 BE)
(100 BE)
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Material 1
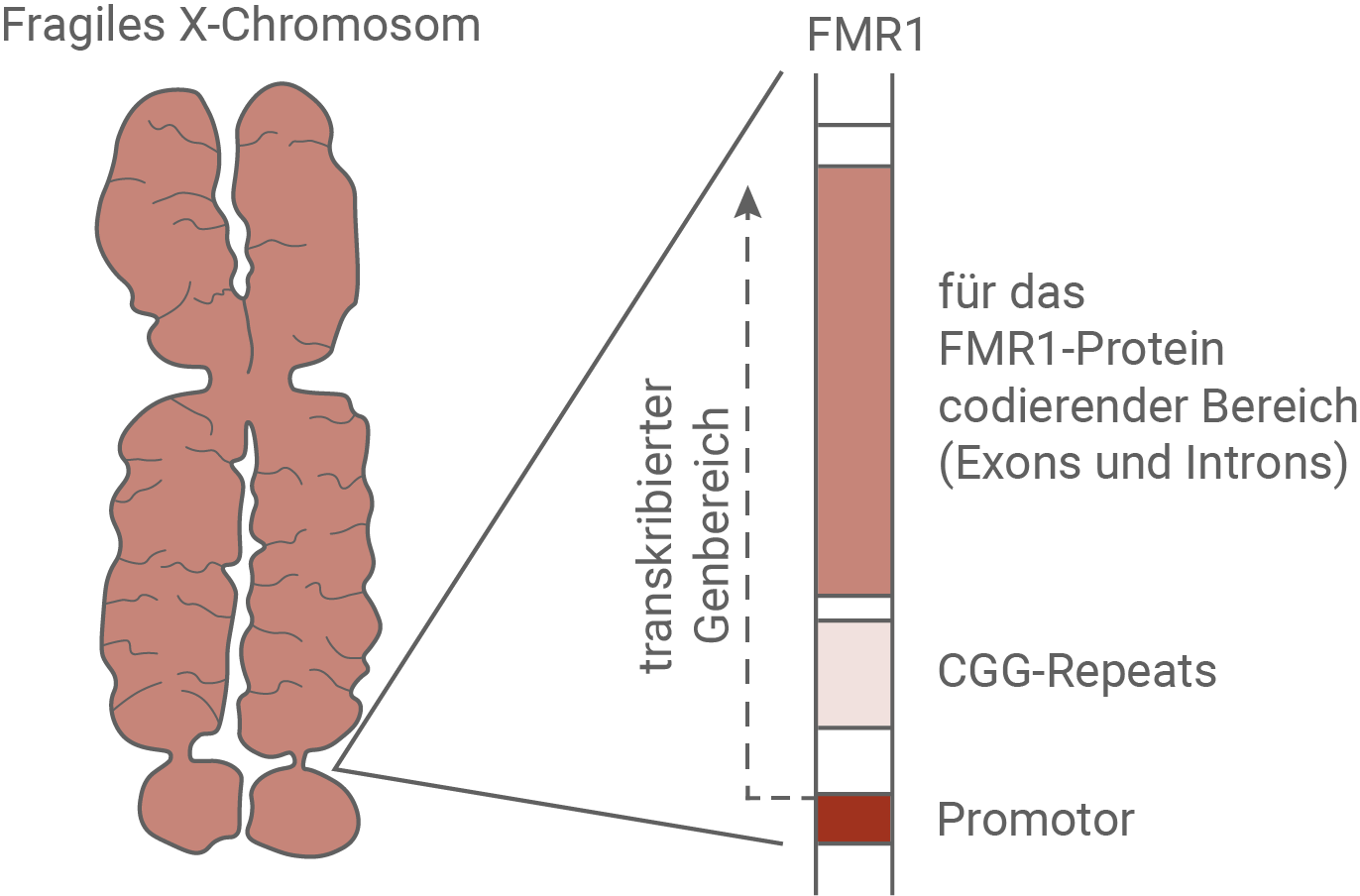
Chromosomen bei Fragilem-X-Syndrom (Fall 1)
Chromosomenuntersuchungen zeigen bei Patienten mit Fragilem-X-Syndrom ein X-Chromosom, das am Ende des längeren Arms eine fragil (brüchig) erscheinende Region aufweist. Die betroffene Region beinhaltet das Gen FMR1, das für das FMR1-Protein (FMRP) codiert (siehe Abbildung). Die Funktionsfähigkeit dieses Proteins spielt eine wichtige Rolle bei der Ausprägung des Fragilen-X-Syndroms.Fragiles X-Chromosom und Aufbau des FMR1-Gens
Material 2
Vererbung des Fragilen-X-Syndroms (Fall 1)
Der Erbgang des Fragilen-X-Syndroms zeigt einige Besonderheiten, da es sowohl gesunde männliche als auch gesunde weibliche Überträger/-innen gibt (siehe Abbildung 2.1). In nahezu allen Fällen wird die Krankheit durch eine Vervielfältigung von CGG-Repeats (CGG-Wiederholungen) im FMR1-Gen hervorgerufen. Auf dem X-Chromosom nicht betroffener Personen befindet sich ein Abschnitt im FMR1-Gen, in dem sich das Basentriplett CGG 5- bis 58-mal wiederholt, in den meisten Fällen 29- oder 30-mal (siehe Abbildung in Material 1). Die Überträger/-innen besitzen entweder ein verändertes, prämutiertes Gen (59-200 CGG-Repeats) oder eine Vollmutation (über 200 CGG-Repeats). Die Vollmutation führt zum Ausbruch der Erkrankung, d. h. zur Ausprägung des Merkmals. In fast allen vollmutierten Allelen sind sämtliche Cytosine des CGG-Repeat-Bereichs sowie angrenzende DNA-Abschnitte bis in den Promotorbereich methyliert.Abb. 2.1: Familienstammbaum mit Fragilem-X-Syndrom
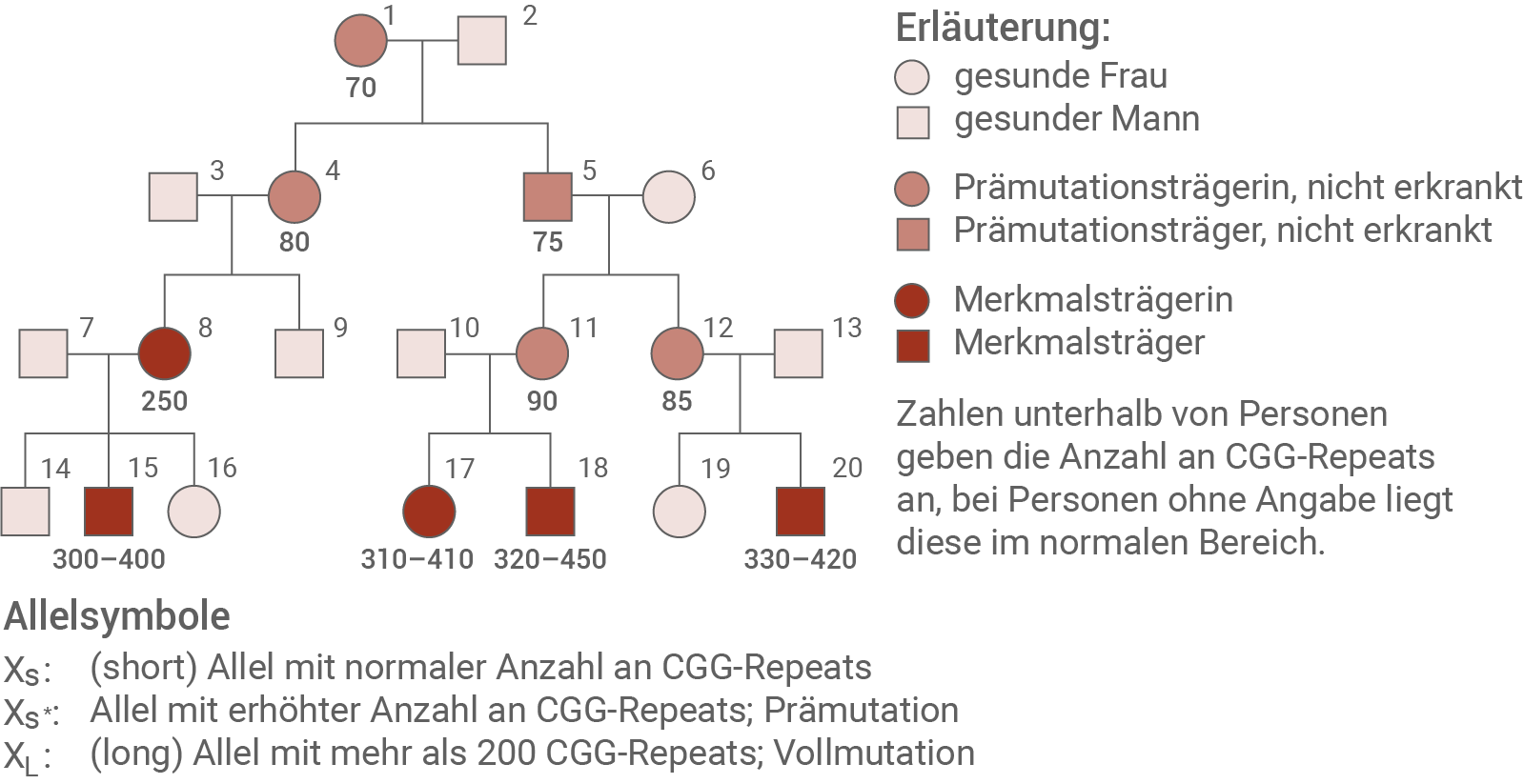
Abb. 2.2: Risiko [%] der Verlängerung eines prämutierten CGG-Repeats in den Vollmutationsbereich
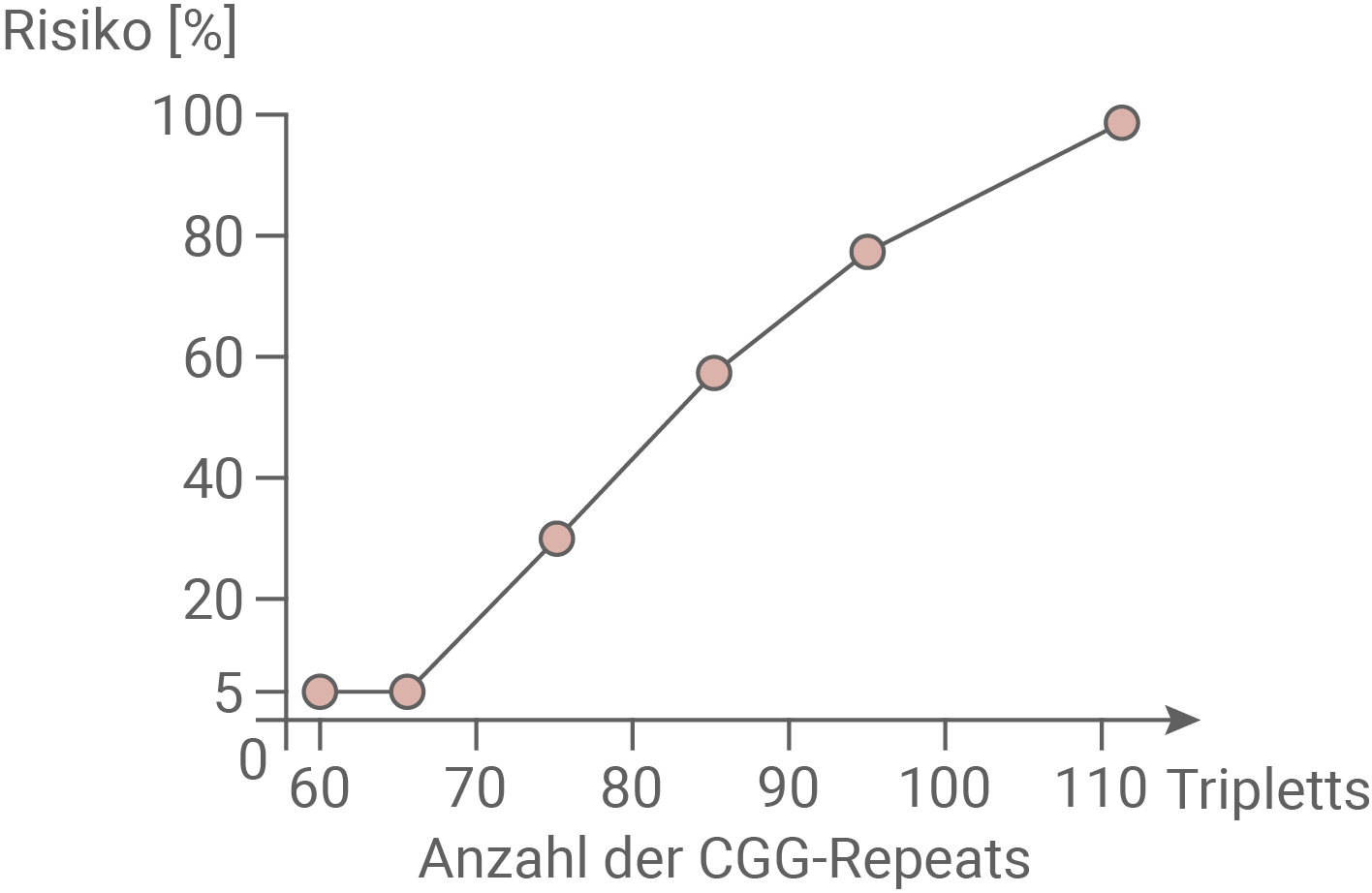
Abb. 2.3: Molekulargenetischer Nachweis von Mutationen bei Fragilem-X-Syndrom
Für den molekulargenetischen Nachweis eines normalen, prämutierten oder vollmutierten Allels wird die aus Blut gewonnene DNA mithilfe eines Restriktionsenzyms geschnitten. Mittels PCR wird das Restriktionsfragment, auf dem die CGG-Repeats des FMR1-Gens liegen, gezielt vervielfältigt und durch Gelelektrophorese analysiert.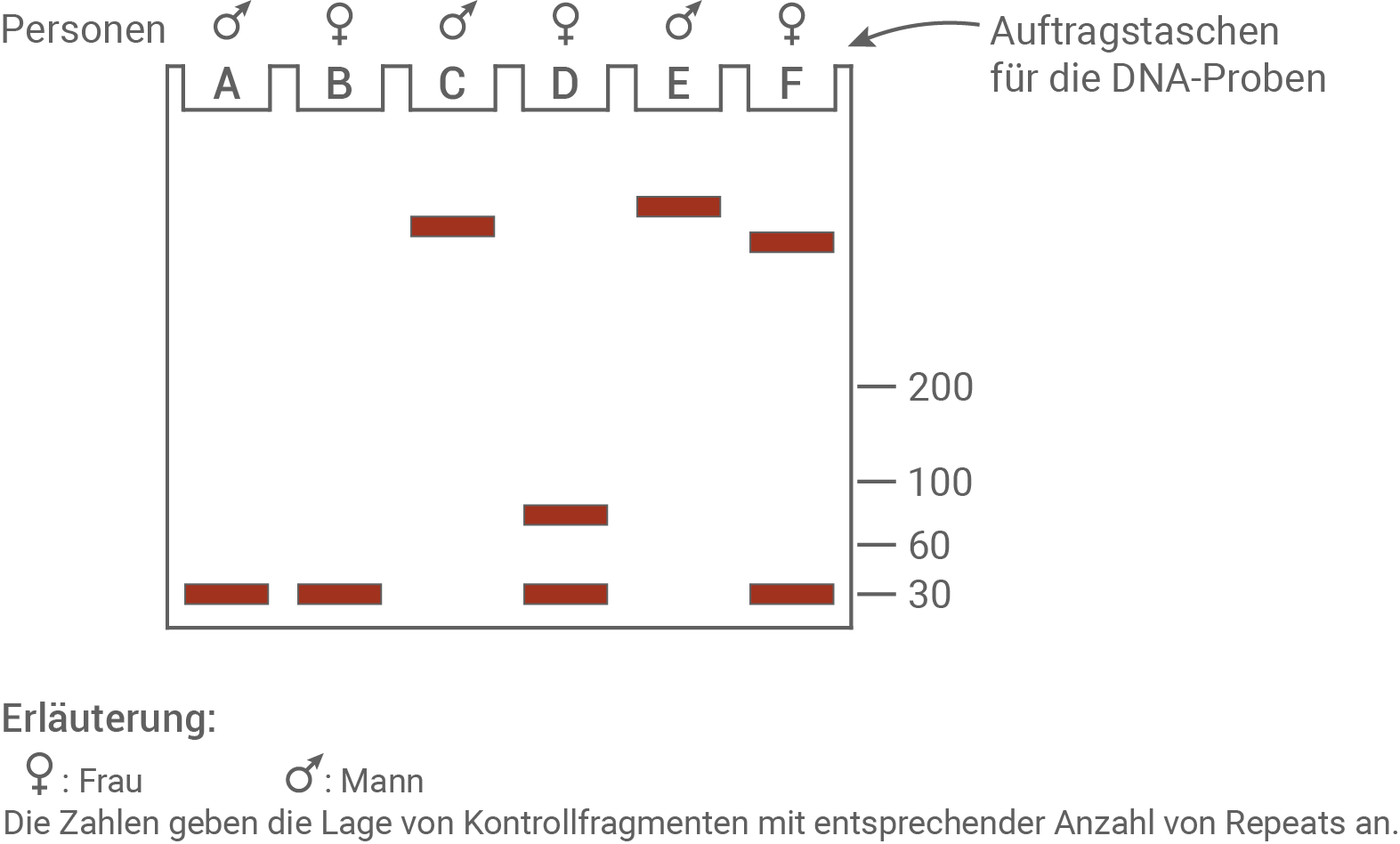
Material 3
Fragiles-X-Syndrom ohne fragiles X-Chromosom (Fall 2)
Bei einigen wenigen von mehreren tausend untersuchten Patienten/-innen, die die typischen Symptome des Fragilen-X-Syndroms, wie Intelligenzminderung und Entwicklungsverzögerung, zeigen, lässt sich keine Vermehrung der CGG-Repeats nachweisen. Wohl aber zeigten diese Personen Veränderungen im FMR1-Protein (FMRP, Abb. 3.3). Diese Entdeckung lieferte neue Ansatzpunkte hinsichtlich der Aufklärung der zellulären Funktionen des FMR1-Proteins. Das FMRP bindet an mRNA und reguliert auf diese Weise insbesondere in Nervenzellen die Translation von Proteinen, die eine wichtige Rolle für die natürliche Funktion der Nervenzellen spielen. FMRP verfügt über drei stabil gefaltete Bereiche (KH1, KH2 und RGG), die an mRNA binden können. Zudem verfügt FMRP über ein sogenanntes nukleäres Lokalisationssignal (NLS) und ein nukleäres Exportsignal (NES) für den Transport in den und aus dem Zellkern.Abb. 3.1: Ausschnitt aus dem nicht-codogenen Strang des FMR1-Gens einer gesunden Person und eines Patienten mit Fragilem-X-Syndrom
Bei einem Patienten mit Fragilem-X-Syndrom, dessen Eltern eine normale Anzahl der CGG-Repeats aufwiesen, entdeckten Wissenschaftler durch molekulargenetische Untersuchung folgende Veränderung der Nukleotidsequenz innerhalb des FMR1-Gens:
Abb. 3.2: Die Code-Sonne der mRNA

Abb. 3.3: Schematische Darstellung von FMRP

Material 4
Symptome des Fragilen-X-Syndroms auf neuronaler Ebene
Während unserer Entwicklung verbinden sich Milliarden von Nervenzellen zu funktionalen Netzwerken, die es dem menschlichen Gehirn erlauben, verschiedene Aufgaben zu bewältigen. Jede Nervenzelle bildet dabei Synapsen aus, die heranreifen, bis sie ihre volle Funktionsfähigkeit erreichen. Beim Fragilen-X-Syndrom zeigen sich vor allem Defizite auf kognitiver Ebene. Betroffene Kinder neigen neben einer Intelligenzminderung zu Aufmerksamkeitsdefiziten, hyperaktivem Verhalten sowie einer vermehrten Gedächtnisauslöschung. Ende der 1990er-Jahre entdeckten Forscher, dass besondere Glutamatrezeptoren, die mGluR5-Rezeptoren, in solchen Gehirnbereichen zu finden sind, die bei der Regulation von Emotionen, Motivation und Gedächtnisbildung eine Rolle spielen. Diese Glutamat führenden Synapsen wirken hier zusammen mit AMPA-Rezeptoren, die Liganden-gesteuerte Ionenkanäle bilden und eine schnelle exzitatorische Erregungsübertragung ermöglichen. Im Tierexperiment mit Modellorganismen, den sogenannten FMR1-Knockout-Mäusen, bei denen das FMR1-Gen entfernt wurde, konnte eine erhöhte Wirkung von Glutamat an Nervenzellen in einer für das Lernen und Erinnern sehr wichtigen Hirnregion, dem Hippocampus, festgestellt werden. Die Zusammenhänge zeigt die folgende Abbildung.Abb. 4.1: Schema der Funktion des FMRP an einer Glutamat führenden Synapse
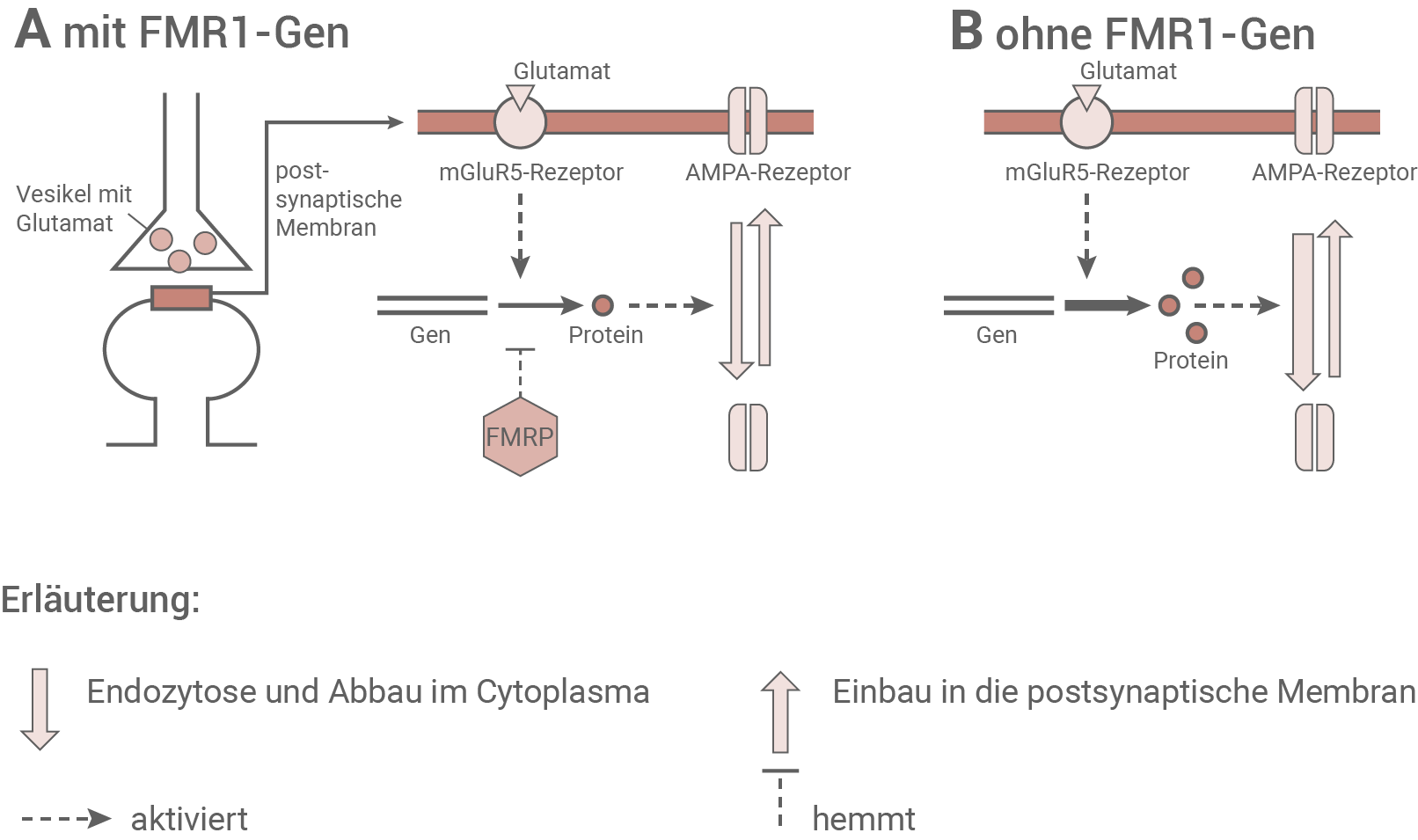
Material 5
Neuronale Plastizität – funktionelle Plastizität
Neuronale Plastizität ist die Fähigkeit des Gehirns, sich in Abhängigkeit seiner Nutzung zu verändern und damit seine Struktur und Organisation veränderten biologischen Anforderungen anzupassen. Dabei bedeutet funktionelle Plastizität, dass sich die Stärke der synaptischen Übertragung aktivitätsabhängig ändert. Die zunehmende Stimulation afferenter Fasern führt zu lang anhaltenden Veränderungen der synaptischen Übertragung im Sinne einer Langzeitpotenzierung („long-term potentiation“, LTP). Für die Auslösung dieser Verstärkung spielen an vielen Synapsen die Bildung und Aktivität der AMPA-Rezeptoren eine Rolle. Eine verringerte Stimulation hingegen führt zu einer Langzeitdepression („Iong-term depression“, LTD), bei der die Übertragungseffizienz vermindert wird. Bei allen diesen Prozessen ist an der Postsynapse insbesondere der Botenstoff Glutamat von entscheidender Bedeutung. Langzeitpotenzierung und Langzeitdepression spielen somit eine wesentliche Rolle bei Lernen und Gedächtnisbildung im Hippocampus.Material 6
Neuronale Plastizität – strukturelle Plastizität
Auf struktureller Ebene bewirkt Langzeitpotenzierung den Umbau und Aufbau neuer Synapsen. Dabei können sich bereits vorhandene Synapsen aktivitätsabhängig teilen, um eine neue Synapse zu bilden, oder die Bildung ganz neuer synaptischer Kontakte anregen. Umgekehrt führt der gegensteuernde Mechanismus, die Langzeitdepression, dazu, dass nicht oder weniger genutzte Synapsen abgebaut werden und so die Übertragungseffizienz vermindert wird. Ganze Dendriten- oder Axonäste können auf diese Weise neu ausgestreckt oder zurückgezogen werden.Material 7
Neuronale Morphologie
Bei Personen mit Fragilem-X-Syndrom besitzen die Dendriten der Nervenzellen – speziell deren faden- oder pilzartigen Fortsätze, die Dornen – eine charakteristische Struktur. Die Struktur der Dornen besitzt eine große Variabilität. Nur an reifen Dornen werden synaptische Kontakte ausgebildet, dies gilt unter anderem auch für die Glutamat führenden Synapsen. Um die Auswirkungen eines Mangels an FMRP auf die dendritische Struktur zu untersuchen, führten Forscher eine Reihe von Studien durch, bei denen Gehirnzellen sowohl von verstorbenen Patienten mit Fragilem X-Syndrom als auch von FMR1-Knockout-Mäusen verwendet wurden. Bei beiden Untersuchungen wurde jeweils auch eine Kontrollgruppe betrachtet.Dornen bei Personen mit Fragilem-X-Syndrom und Kontrollpersonen
Abb. 7.1: Mikroskopisches Bild eines Dendriten mit Dornen

Abb. 7.2: Verschiedene Strukturen von Dornen (Schema)
Abb. 7.3: Anteil verschiedener Dornenstrukturen im Gehirn
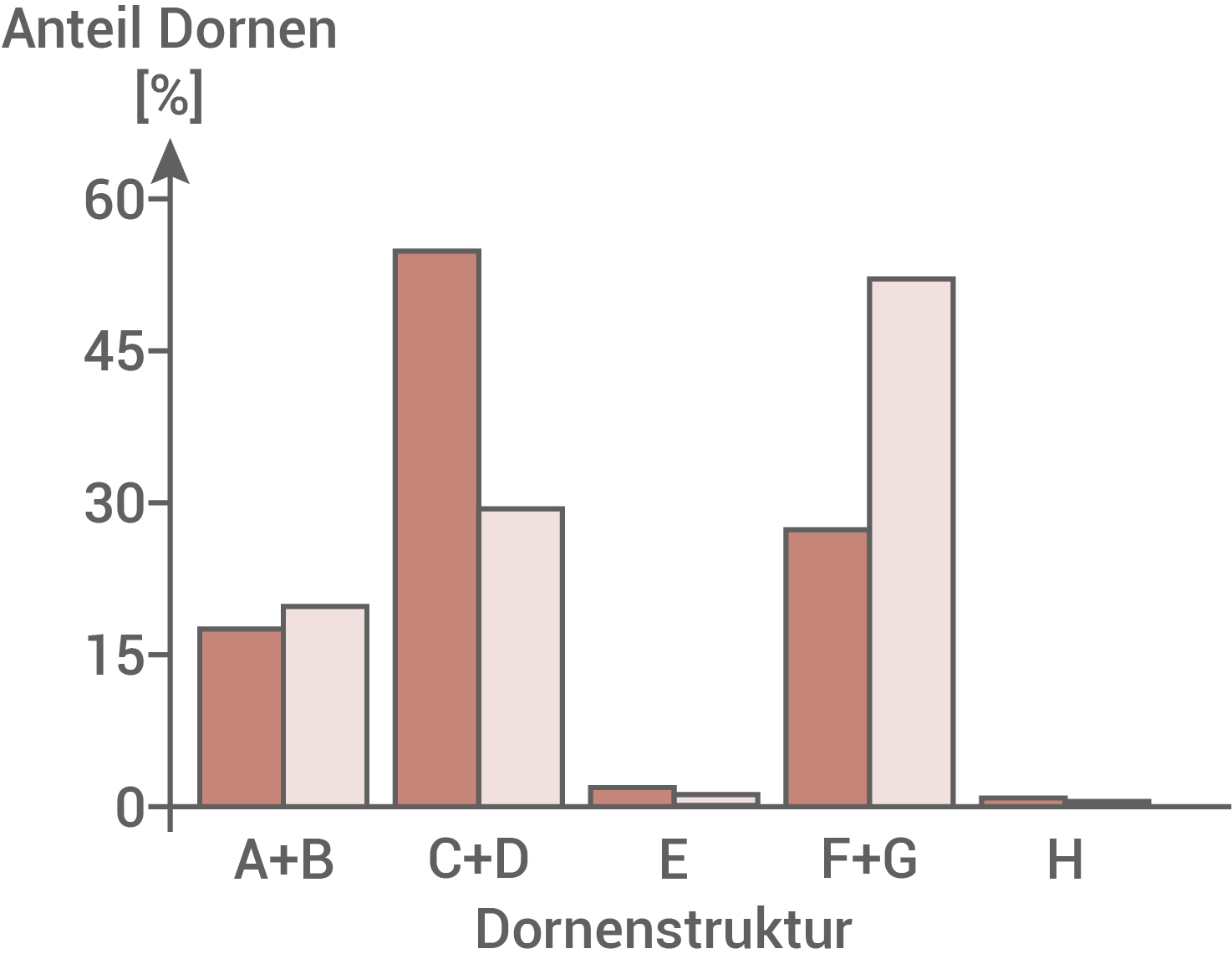
Abb. 7.4: Anteil verschiedener Dornenlängen im Gehirn
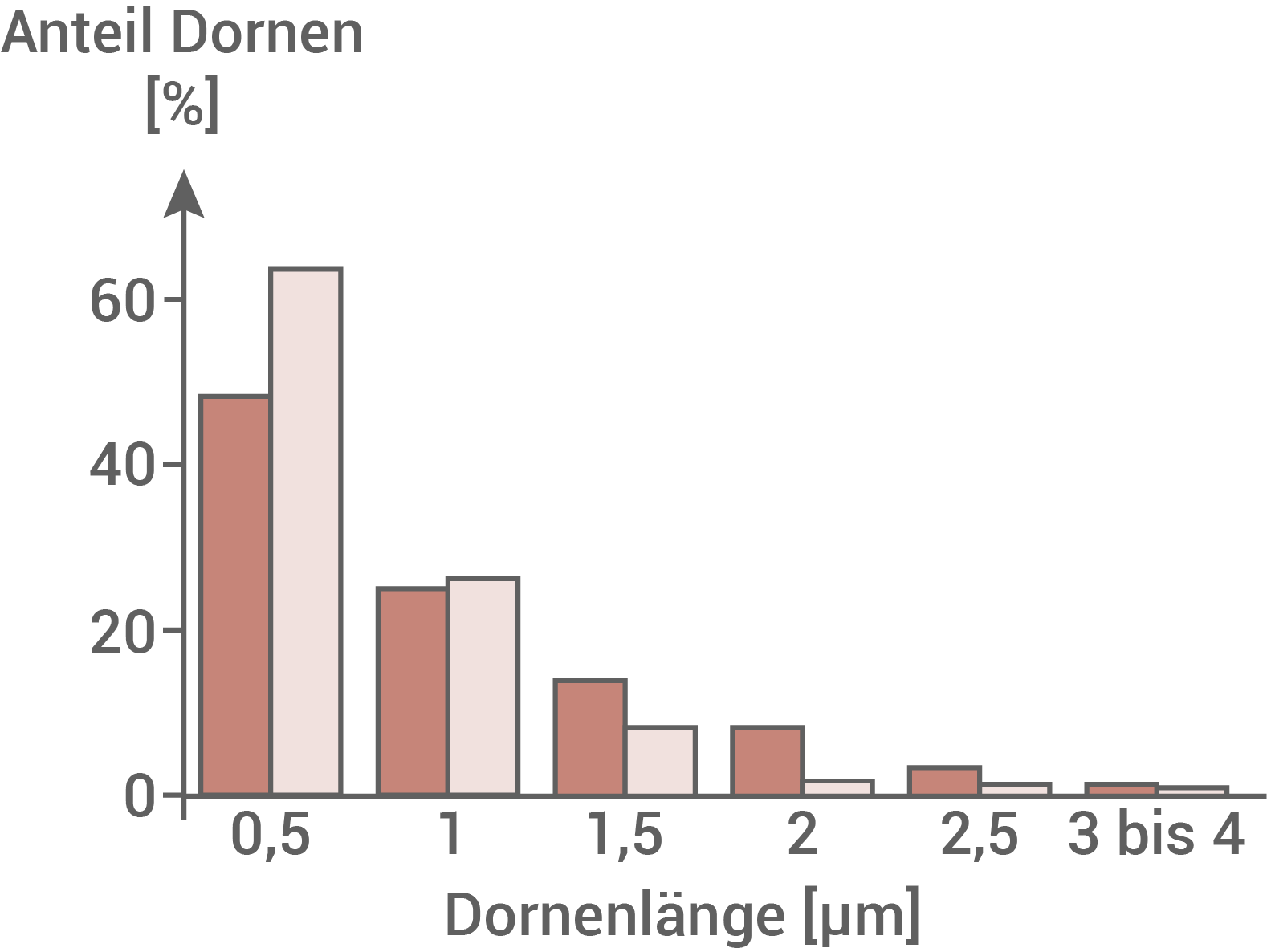

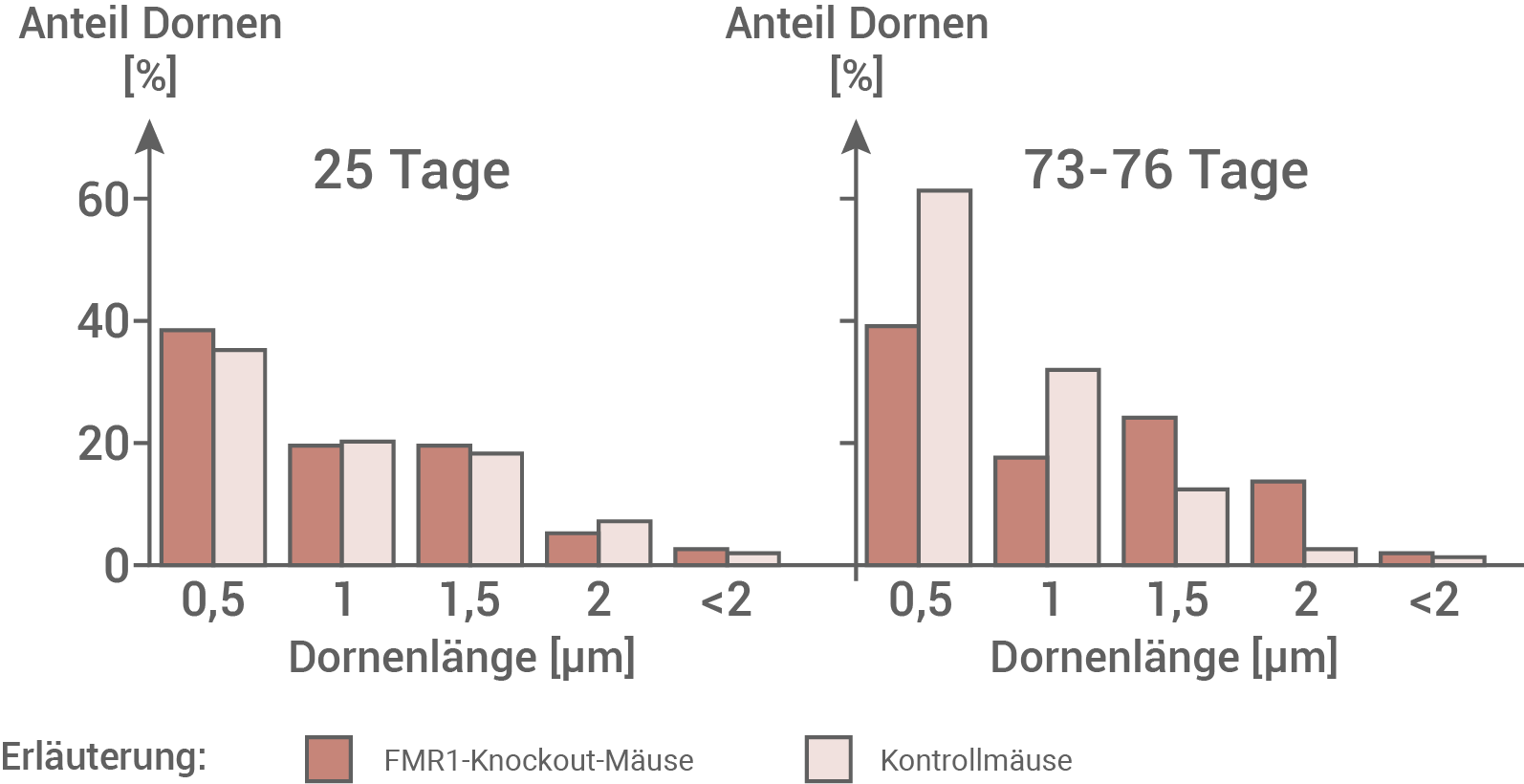
Abb. 7.5: Anteil verschiedener Dornlängen im Gehirn von Mäusen unterschiedlichen Alters
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?
1
Vorgänge der Transkription und der Reifung der mRNA bei Eukaryoten sowie der Einfluss der DNA-Methylierung auf die Proteinbiosynthese:
- Transkription:
Der DNA-Strang wird von der RNA-Polymerase an der Promotorregion geöffnet, wobei die Wasserstoffbrücken zwischen den einzelnen Basen aufgelöst werden. An den codogenen Strang lagern sich nun im Zellkern vorkommende Nukleotide komplementär an, und werden von der RNA-Polymerase in 5'3' Richtung miteinander verknüpft. Eine definierte Basenabfolge, das Polyadenylierungssignal, vermittelt das Ende der Transkription. Die noch nicht prozessierte mRNA wird daraufhin freigesetzt.
- Reifung der mRNA:
Die prä-mRNA enthält Passagen, die nicht für ein bestimmtes Gen codieren. Diese Passagen werden Introns genannt, und müssen im Zuge der Prozessierung ausgespleißt werden, sodass am Ende nur noch die codierenden Exons in der mRNA enthalten sind. An das 5'-Ende der prä-mRNA wird außerdem eine Cap-Struktur angefügt. Am 3'-Ende wird der Poly-A-Schwanz synthetisiert, der die mRNA gemeinsam mit der Kappe vor frühzeitigem Abbau schützen soll. - Einfluss der DNA-Methylierung:
Die DNA-Methylierung zeigt bei Eukaryoten nicht transkribierte Bereiche der DNA an. Dabei blockieren Methylgruppen die betroffene DNA-Sequenz, sodass diese für die RNA-Polymerase unzugänglich werden. Dadurch wird die Transkription dieser Bereiche verhindert.
2
Bestimmung der Genotypen:
- Person 1:
oder
- Person 2:
- Person 5:
- Person 6:
- Person 11:
- Person 17:
- Person 18:
3
Prinzip der Gelelektrophorese:
Das Verfahren der Gelelektrophorese dient der Unterscheidung geladener Teilchen (wie zum Beispiel DNA-Fragmente) nach ihrer Größe. Durch eine anschließende Färbung des Gels können diese Fragmente sichtbar gemacht werden. Die Gelelektrophorese besteht aus einer Kammer, in der sich ein Agarose-Gel befindet. Das Gel ist strukturell wie ein engmaschiges Netz aufgebaut, wodurch größere Fragmente langsamer wandern als kleinere. Auf der Kathodenseite des Gels werden die zu untersuchenden Proben gemeinsam mit einem Ladepuffer in dafür vorgesehene Taschen im Gel gegeben. In der Regel wird in eine äußere Tasche ein Größenmarker gegeben, um die DNA-Fragmente später zuordnen zu können. Im Anschluss wird eine elektrische Spannung angelegt, und die Fragmente beginnen zu wandern. Da die DNA polar und durch ihr Phosphatrückgrat negativ geladen ist, wandert sie im Gel von der Kathode in Richtung Anode. Nach einer definierten Zeitspanne, wird das Gel von der Spannungsquelle getrennt, und mit einem Farbstoff eingefärbt, der die Banden im UV-Licht sichtbar macht. Dadurch und durch den Vergleich mit dem Größenmarker kann die Länge und Häufigkeit der erhaltenen DNA Fragmente ermittelt werden.
Begründete, exemplarische Zuordnung der Bandenmuster:
- Bandenmuster A: Diese Bande gehört zu einem gesunden Mann, da nur eine Bande mit einer Länge von 30 Repeats zu erkennen ist, was der GCC-Repeat-Menge einer gesunden Person entspricht. Dafür würden die Personen 2, 3, 7, 9, 13 und 14 infrage kommen.
- Bandenmuster B: Diese Bande gehört zu einer gesunden Frau, da nur eine Bande mit S-Fragmenten vorliegt. Dazu würden die Personen 6, 16, und 19 passen.
- Bandenmuster C und E: Hier liegen die Banden zweier erkrankter Männer vor. Die Banden haben eine Länge von über 200 Repeats. Es liegen demnach L-Fragmente vor und die Mutation kann als Vollmutation gedeutet werden. Dabei zeigt die Bande E etwas mehr Repeats an, als die Bande C. Die hier infrage kommenden Personen sind 15 und 20, 20 und 18 oder 15 und 18.
- Bandenmuster D: Die Bande stammt von einer weiblichen Person mit Prämutation. Die Person muss dabei heterozygot sein, da eine Bande bei 30 Repeats (gesundes X-Chromosom) und eine bei knapp unter 100 (Prämutation des anderen X-Chromosoms) zu erkennen ist. Dafür kommen die Personen 1, 4, 11 und 12 infrage.
- Bandenmuster F: Das Bandenmuster ist einer Frau mit Vollmutation zuzuordnen. Sie ist heterozygot, da das Bandenmuster ein S-Fragment (normale Länge) und ein L-Fragment (vollmutiert) zeigt. Hierfür kommen die Personen 8 und 17 infrage.
4
Aminosäuresequenz des FMR1-Protein-Ausschnitts einer gesunden Person und eines Patienten mit Fragilem-X-Syndrom:
- Gesunde Person: Lys–Leu–Ile–Gln–Glu
- Patient: Lys–Leu–Asn–Gln–Glu
5
Auswirkung der jeweiligen Mutation auf Bildung und Struktur des FMRP-Proteins (FMRP) für beide Formen des Fragilen-X-Chromosoms:
- Fall 1: Die Krankheit bricht aus, wenn ein Patient eine Mutation mit über 200 Wiederholungen der CGG-Tripletts aufweist. Bei fast allen vollmutierten Allelen sind sämtliche Cytosine inklusive Promotorbereich methyliert. Dies hat zur Folge, dass dieser Abschnitt nicht transkribiert werden kann.
- Fall 2: In wenigen Fällen ist die Grundlage der Krankheit keine übermäßige Repeatanzahl des CGG-Tripletts, sondern eine Punktmutation in der zweiten Base in Position 304 der Aminosäuresequenz. Durch diese Mutation wird nicht wie bei einer gesunden Person die Aminosäure Leucin eingebaut, sondern Asparagin. Die Missense-Mutation hat zur Folge, dass das Protein durch die veränderte Primärstruktur eine andere Konformation einnimmt, und seine Funktion dadurch beeinträchtigt ist.
6
Beschreibung der Erregungsübertragung einer erregenden Acetylcholin führenden Synapse:
Eine Acetylcholin führende erregende Synapse ist für die Weiterleitung von Reizen, beispielsweise zwischen zwei Nervenzellen, zuständig. Am Ende eines Axons befindet sich die Synapse, die über die präsynaptische Membran mit dem synaptischen Spalt in Kontakt steht. Als synaptischen Spalt bezeichnet man eine etwa 20–30 nm breite Lücke zwischen zwei Synapsen. Erreicht ein Aktionspotenzial die Präsynapse, so öffnen sich in deren Membran spannungsabhängige Calcium-Ionenkanäle, und es kommt zu einem Einstrom positiv geladener Calciumionen. Die dadurch hervorgerufene erhöhte Calcium-Ionenkonzentration in der Synapse bewirkt die Wanderung der mit dem Transmittermolekül Acetylcholin gefüllten synaptischen Bläschen. Diese bewegen sich in Richtung der präsynaptischen Membran, wo sie schließlich mit ihr verschmelzen. Durch Exocytose wird der Transmitterstoff in den synaptischen Spalt entlassen, und gelangt durch Diffusion bis zur postsynaptischen Membran. In der postsynaptischen Membran befinden sich ligandengesteuerte Natrium-Ionenkanäle. Acetylcholin ist in der Lage, an diese zu binden, wodurch die Öffnung der Natrium-Ionenkanäle induziert wird. Positiv geladene Natriumionen strömen in die Zelle ein, und lösen so eine Depolarisation der postsynaptischen Membran aus. Es entsteht ein EPSP (exzitatorisches postsynaptisches Potenzial). Das Transmittermolekül löst sich von den Rezeptoren in der postsynaptischen Membran, wodurch die Natrium-Ionenkanäle schließen und der Natrium-Ioneneinstrom unterbunden wird. Das Enzym Acetylcholinesterase ist in der Lage, Acetylcholin in seine Grundbestandteile zu spalten. Über Endocytose werden diese nun wieder in die Präsynapse aufgenommen, zu Acetylcholin zusammen gesetzt, und in den synaptischen Bläschen gespeichert. Calcium-Ionenpumpen transportieren die anfangs eingeströmten Calciumionen unter ATP-Verbrauch wieder aus der Präsynapse hinaus.
Vorgänge an der postsynaptischen Membran, die zur Entstehung eines IPSP führen:
Inhibitorische postsynaptische Potenziale entstehen nur an hemmenden Synapsen. Der Unterschied zu erregenden Synapsen liegt dabei in Struktur und Aufbau der postsynaptischen Membran. Bindet das Transmittermolekül an Rezeptoren in der postsynaptischen Membran, so löst das die Öffnung transmittergesteuerter Kalium-Ionenkanäle oder Chlorid-Ionenkanäle aus. Es strömen nun gemäß dem Konzentrationsgefälle entweder Kaliumionen aus der Zelle aus, oder Chloridionen in die Zelle hinein. Dies führt zu einer Hyperpolarisation der postsynaptischen Membran. Die Erregung wird dadurch inhibiert.
7
Funktionsweise der Glutamat führenden Synapse und Folgen der beiden Mutationsformen im FMR1-Gen für die Funktion dieser Synapsen:
Betrachtet man das Schema zur Funktion einer Glutamat-führenden Synapse, so lässt sich erkennen, dass die identischen mGluR5-Rezeptoren, wenn sie mit Glutamat besetzt sind, die Genexpression aktivieren. Die dadurch entstehenden Proteine sind für den vermehrten Einbau von excitatorisch wirksamen AMPA-Rezeptoren in die postsynaptischen Membran, oder deren Abbau durch Endocytose ins Cytoplasma zuständig. Die Stärke des Abbaus bzw. Einbaus ist dabei von der Menge des gebildeten Proteins abhängig. Bei einer hohen Proteinkonzentration überwiegt der Abbau der Rezeptoren. Das FMR1-Protein hemmt die Proteinbiosynthese, verringert dadurch die Proteinbiosynthese und es findet vermeht ein Einbau der AMPA-Rezeptoren statt. Bei Patienten mit Fragilem-X-Syndrom fehlt FMRP oder ist in seiner Funktion beeinträchtigt. Dadurch kann die Proteinbiosynthese nicht mehr reguliert werden, es wird viel Protein gebildet, und es findet ein verstärkter Abbau der AMPA-Rezeptoren statt. Infolgedessen kommt es bei der Erregungsweiterleitung zu einem abgeschwächten EPSP und die Effektivität der Erregungsübertragung ist verringert.
Bei dem Fragilem-X-Syndrom auftretenden Symptome und Deutung unter neurophysiologischen Gesichtspunkten:
Durch das fehlende FMR1-Protein werden AMPA-Rezeptoren aus der postsynaptischen Membran abgebaut. Dies führt wiederum zu einer dauerhaften Abschwächung des EPSP. Die ineffiziente Erregungsübertragung in diesen Hirnregionen beeinträchtigt die Regulation von Emotionen, und kann die Motivation sowie die Gedächtnisleistung schwächen. Durch neuronale Plastizität können Langzeitdepressionen und kognitive Defizite entstehen.
8
Wirkungsweise für ein im synaptischen Spalt wirkendes Medikament, welches die Symptome der Betroffenen mildern könnte:
Die Symptome der Patienten mit fragilem X-Syndrom beruhen auf der überschwelligen Wirkung von Glutamat, wobei die Regulation des Transmittermoleküls gestört ist. Ein Medikament, welches im synaptischen Spalt wirksam ist, könnte Glutamat reversibel binden und dadurch die Glutamatkonzentration im synaptischen Spalt herabsetzen. Alternativ könnte ein Medikament zum Einsatz kommen, welches die mGluR5-Rezeptoren in der postsynaptischen Membran besetzt, sodass diese für Glutamat nicht mehr zugänglich sind. Um diesen Effekt zu erzielen, muss der Wirkstoff als Antagonist zu Glutamat wirken, und eine ähnliche Raumstruktur aufweisen, um die Rezeptorbindestelle effektiv zu blockieren.
9
Zusammenfassung der Untersuchungsergebnisse von Abbildung 7.3 und 7.4:
Durch den Vergleich der mikroskopischen Bilder in Abbildung 7.1 kann gezeigt werden, dass Patienten mit Fragilem-X-Chromosom eine veränderte Form der Dornen an den Dendriten aufweisen. Ein Mangel an funktionsfähigem FMRP führt demnach zu einem veränderten prozentualen Anteil der Dornenstruktur und der Dornenlängen. Abbildung 7.3 ist zu entnehmen, dass erkrankte Personen prozentual mehr Dornen der Struktur C und weniger Dornen der Struktur F+G aufweisen, als gesunde Personen. Tendenziell sind die Dornen der Patienten mit Fragilem-X-Syndrom eher verlängert und damit unreif, wie den Abbildungen 7.2 und 7.4 zu entnehmen ist.
Analyse des Untersuchungsergebnisses von Abbildung 7.5:
In dem Experiment lässt sich zeigen, dass sich der prozentuale Anteil an Dornen bei Patienten und gesunden Personen nicht erheblich unterscheidet. Erst ab einem Alter von 73–76 Tagen zeigt sich eine Divergenz hinsichtlich der Dornenlänge und dem prozentualen Auftreten von Dornen einer bestimmten Länge. So liegt der Anteil der reifen Dornen bei Mäusen der Kontrollgruppe bei 65 %, während bei FMR1-Knockout Mäusen nur 40 % der Dornen reif sind. Dies bedeutet, dass sich die Krankheit erst nach den ersten 25 Lebenstagen erkennen lässt, und die Reifung durch die fehlende Wirkung von FMRP bei den Knockout-Mäusen eingeschränkt ist.
Deutung der Ergebnisse hinsichtlich der Gedächtnisleistung:
Nur die reifen bzw. pilzförmigen Dornen der Dendriten können synaptische Kontakte ausgebildet werden. Dies gilt auch für die betrachteten Glutamat führenden Synapsen. Es ist also zu vermuten, dass diese Synapsen bei Patienten mit Fragilem-X-Syndrom nur in geringem Maße ausgeprägt werden. Dies führt wiederum zu einer strukturellen Langzeitdepression, bei der die Übertragungseffizienz der Neuronen vermindert ist, und eine geringere Gedächtnisleistung der Betroffenen zur Folge hat.