Vorschlag B – Epilepsie
Hinweis: Von den Vorschlägen A und B soll in der Prüfung nur einer bearbeitet werden.
Epilepsie – Gewitter im Gehirn
Im menschlichen Gehirn sind über eine Billion Neuronen zu einem Organ verschaltet, das unsere körperlichen und geistigen Funktionen steuert. Kommt es zu Mutationen in den Genen derjenigen Proteine, welche die Entwicklung des jungen Gehirns steuern, kann dies Fehlbildungen zur Folge haben mit zum Teil schwerwiegenden Auswirkungen auf physiologische und kognitive Prozesse. Eine mögliche Folge sind beispielsweise epileptische Anfälle, die durch nicht kontrollierte Erregungen von Neuronengruppen im Gehirn ausgelöst werden und mit Bewusstseinsstörungen oder auch Krampfzuständen der Muskulatur einhergehen.Synaptische Übertragung und Verschaltung von Neuronen
1
Beschreibe jeweils die Entstehung eines excitatorischen Potenzials (EPSP) sowie eines inhibitorischen Potenzials (IPSP) an der postsynaptischen Membran eines Neurons. Gib jeweils eine Definition für die Begriffe zeitliche Summation und räumliche Summation an.
(11 BE)
2
Erkläre die Ergebnisse der Messungen an den Messstellen 1 und 2. Leite die Bedeutung von Renshaw-Neuronen für die Steuerung der Muskelkontraktion bei der Aktivierung von Motoneuronen durch absteigende Nervenbahnen her. (Material 1)
(14 BE)
Wirkung von Antiepileptika
3
Analysiere die Ergebnisse der Untersuchungen. (Material 2)
(6 BE)
4
Stelle die verschiedenen Wirkorte des Antiepileptikums Topiramat an Glutamat führenden Synapsen mit Hilfe einer Skizze dar. Erläutere ausgehend von diesen Wirkorten den Einfluss von Topiramat auf die Erregungsübertragung der Synapsen. (Material 3)
(14 BE)
Molekularbiologische Nachweismethoden für Mutationen
5
Beschreibe den prinzipiellen Ablauf einer Polymerase-Kettenreaktion sowie einer Gelelektrophorese.
(13 BE)
6
Prüfe den Stammbaum der Familie mit einer erblichen Form der Lissenzephalie auf mögliche Vererbungsmodi. Entscheide anhand der auftretenden Symptome, welcher der möglichen Vererbungsmodi wahrscheinlich vorliegt. (Material 4).
(18 BE)
7
Gib für den DNA-Ausschnitt des DCX-Gens einer an Lissenzephalie erkrankten Person die entsprechende Aminosäuresequenz des Proteins Doublecortin an. Bestimme die Mutationsform und erkläre mögliche Folgen für die Gehirnentwicklung. (Material 4 und 5)
(9 BE)
8
Beschreibe die Unterschiede in den Ergebnissen der Gelelektrophorese im Vergleich und deute den Befund des Lissenzephalie-Patienten. (Material 5 und 6)
(15 BE)
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Material 1
Verschaltung von Neuronen
Im Rückenmark von Wirbeltieren befinden sich Motoneurone, die Skelettmuskelfasern innervieren und somit sowohl Bewegungen ermöglichen als auch die muskuläre Spannung steuern. Sie enthalten den Transmitter Acetylcholin. Alle von einem bestimmten Motoneuron innervierten Muskelzellen und das zugehörige Motoneuron selbst bilden eine motorische Einheit. Eine motorische Einheit kann einige wenige bis zu mehreren tausend Muskelzellen umfassen. Je kleiner die motorischen Einheiten eines Muskels sind, desto feiner können Bewegungen abgestuft werden. Alle motorischen Einheiten eines Muskels tragen gemeinsam zu seiner Kontraktionskraft bei.Ein einzelnes Neuron kann tausende präsynaptischer Endigungen von anderen Neuronen besitzen, von denen ein Teil erregend, andere hemmend wirken. Darüber hinaus können sich Neurone verzweigen und über diese Verzweigungen, Kollateralen genannt, andere Neurone innervieren.
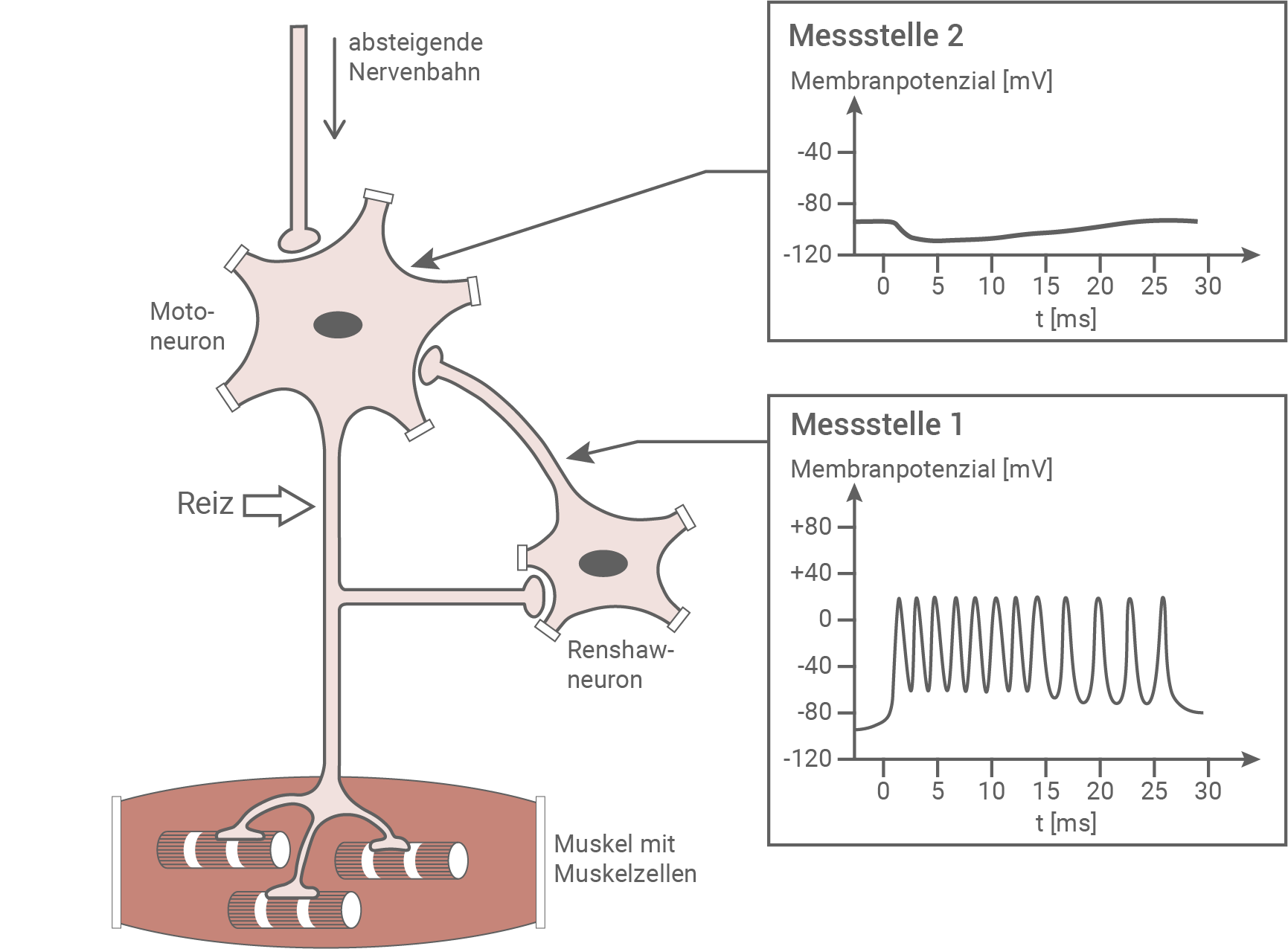
Im Rückenmark sind Motoneurone über Kollateralen mit spezialisierten Interneuronen verschaltet, die nach ihrem Entdecker als Renshaw-Zellen bezeichnet werden. Sie reagieren so empfindlich auf Signale von Motoneuronen, dass ein einziges Aktionspotential eines Motoneurons ausreicht, um in einem Renshaw-Neuron eine Serie von Potentialänderungen auszulösen. Die folgende Abbildung zeigt ein Motoneuron, das einen Muskel innerviert. Das Axon des Motoneurons ist über eine Kollaterale mit einem Renshaw-Neuron verbunden. Das Renshaw-Neuron schüttet den Transmitter Glycin aus.
In einem Experiment wird das Axon des Motoneurons künstlich gereizt, und die entstehenden Potenziale werden am Axon der Renshaw-Zelle (Messstelle 1) sowie am Soma des Motoneurons (Messstelle 2) abgeleitet.
Verschaltung eines Motoneurons mit einem Renshaw-Neuron und experimentelle Ableitung von Potenzialen (vereinfachte Darstellung)
Roger Eckert, David Randall, Warren Burggren, Kathleen French: Tierphysiologie, Thieme Verlag Stuttgart, 4. Aufl. 2002, S. 470.
Gerhard Thews, Ernst Mutschler, Peter Vaupel: Anatomie Physiologie Pathophysiologie des Menschen, Wissenschaftliche Verlagsgesellschaft mbH Stuttgart 4. Aufl. 1991, S. 410.
Francisco Alvarez, Robert Fyffe: The continuing case for the Renshaw cell, The Journal of Physiology, 584,2007, S. 31-45,2018,
https://www.researchgate.net/publication/6197563_The_continuing_case_for_the_Renshaw_cell
https://www.spektrum.de/lexikon/biologie/motorische-einheit/44141
Material 2
Epilepsie
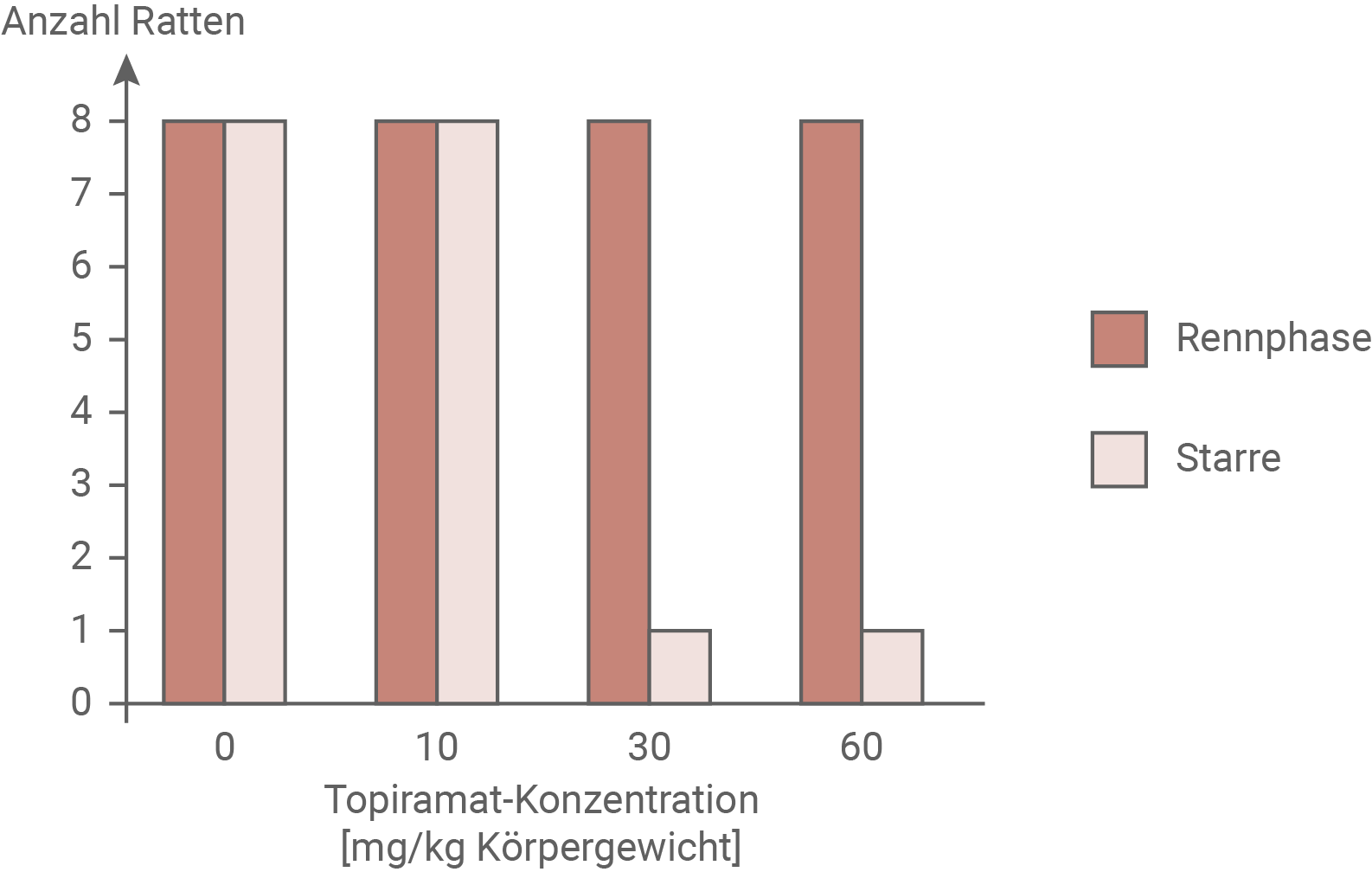
Epilepsie ist die häufigste chronische Erkrankung des zentralen Nervensystems. Nach Angaben der Weltgesundheitsorganisation WHO erleiden zirka fünf Prozent aller Menschen mindestens einmal in ihrem Leben einen epileptischen Anfall. Dabei gerät die neuronale Aktivität außer Kontrolle: Viele Nervenzellen aktivieren sich selbstständig zur gleichen Zeit, was Symptome wie zum Beispiel Bewusstseins- und Wahrnehmungsstörungen, Muskelzuckungen oder Krampfzustände verursachen kann. Diese Entladungen von Nervenzellen können sich über das gesamte Gehirn ausbreiten. Es wird vermutet, dass die mangelnde Koordination von neuronaler Erregung und Hemmung zur Auslösung epileptischer Anfälle beiträgt. Neben einer erblichen Disposition können auch Hirnschädigungen oder angeborene Fehlbildungen des Gehirns Epilepsie verursachen. Um die Wirksamkeit von Medikamenten gegen Epilepsie zu testen, nutzt man aus, dass sich bei bestimmten Stämmen von Ratten epileptische Anfälle durch akustische Reize auslösen lassen. Diese Anfälle werden durch eine Phase intensiven Rennens eingeleitet, die jeweils 10 bis 30 Sekunden andauert. Darauf folgt typischerweise eine Phase der Starre, während der die Ratte in einem muskulären Krampfzustand mit steif ausgestreckten Gliedmaßen unbeweglich verharrt. In einer Serie von Experimenten wurden acht Ratten 60 Minuten nach der Behandlung mit dem Wirkstoff Topiramat einem akustischen Reiz ausgesetzt, der geeignet war, einen epileptischen Anfall auszulösen. Bei verschiedenen Dosierungen von Topiramat wurde untersucht, wie viele der untersuchten Ratten als Reaktion auf einen akustischen Reiz die typische Abfolge eines solchen epileptischen Anfalls mit intensivem Rennen, gefolgt von einem anschließenden Starrezustand, zeigten.Wirkung von Topiramat auf epileptische Anfälle
Epilepsie - Mehr als «nur» Zucken und Schreien, Stiftung MyHandicap,
https://www.myhandicap.ch/gesundheit/koerperliche-behinderung/epilepsie/ (Absatz 1)
https://www.epilepsie-gut-behandeln.de/was-ist-epilepsie (abgerufen am 06. 04. 2020); Marie-Aude Rigoulot, Any Boehrer, Astrid Nehlig: Effects of Topiramate in Two Models of Genetically Determined Generalized Epilepsy, the GAERS and the Audiogenic Wistar AS, Epilepsia 44 (1): 2003, S. 14-19. (Absatz 2-5)
https://www.epilepsie-gut-behandeln.de/was-ist-epilepsie
Material 3
Antiepileptika
Die Behandlung von Patienten mit Epilepsie erfolgt durch die Gabe von anti-epileptischen Medikamenten, sogenannten Antiepileptika. Ihre Wirkung zielt vor allem auf eine Senkung der neuronalen Überaktivität im Gehirn sowie auf das Lösen von Krampfzuständen der Muskulatur ab. Die medikamentöse Behandlung bei Epilepsie ist eine Langzeitbehandlung, die sich über das ganze Leben erstrecken kann. Der Wirkstoff Topiramat, der zur Behandlung von Epilepsie eingesetzt wird, blockiert im zentralen Nervensystem unter anderem spannungsabhängige Calcium-Ionenkanäle an erregenden Synapsen, die den Transmitter Glutamat verwenden. Auch bindet Topiramat an die Glutamat-Bindungsstellen von Rezeptoren in der postsynaptischen Membran, ohne die Ionenkanäle für Natriumionen zu öffnen. Glutamat ist der im ZNS quantitativ am häufigsten vorkommende erregende Neurotransmitter. Material 3 basiert auf:https://www.myhandicap.ch/gesundheit/koerperliche-behinderung/epilepsie/medikamente-beiepilepsie/
https://www.pharmawiki.ch/wiki/index.php?wiki=topiramat
https://neurolab.eu/infos-wissen/wissen/neurotransmitter/glutamat/
Material 4
Lissenzephalie
Lissenzephalie ist eine neurologische Erkrankung, die auf einer Entwicklungsstörung der Nervenzellen in der menschlichen Großhirnrinde beruht. Diese führt, je nach Schweregrad der Erkrankung, dazu, dass die Faltung der Großhirnrinde vollständig fehlt bzw. nur eine grobe Strukturierung vorliegt. Auch kann die Anordnung der Schichten neuronaler Gewebe innerhalb der Hirnrinde verändert sein. Die bei Lissenzephalien beobachteten Fehlbildungen entstehen bereits im Mutterleib während der pränatalen Entwicklung des kindlichen Gehirns. Durch die Fehlbildungen ihres Gehirns leiden an Lissenzephalie erkrankte Menschen oftmals unter geistigen Behinderungen und epileptischen Anfällen. Es werden auch Störungen der motorischen Fähigkeiten beobachtet sowie Beeinträchtigungen beim Schlucken, die in schweren Fällen eine Ernährung über eine Magensonde notwendig machen. Das Spektrum der Beeinträchtigungen reicht von leichten Formen mit epileptischen Anfällen bis zu schwersten Verläufen, die bereits innerhalb der ersten Lebensmonate zum Tod führen können. Eine ursächliche Therapie gibt es zurzeit nicht. Eine häufige Ursache der erblichen Form der Lissenzephalie sind Mutationen im Gen DCX, das das Protein Doublecortin kodiert. Doublecortin wird ausschließlich in den ersten Monaten der frühkindlichen Entwicklung in neuronalen Geweben exprimiert. In sich entwickelnden Neuronen bindet es an die Mikrotubuli des Cytoskeletts und stabilisiert diese. Diese Funktion von Doublecortin spielt eine wichtige Rolle bei der Wanderung und der korrekten Positionierung junger Neurone in der Großhirnrinde.Stammbaum einer Familie mit einer erblichen Form der Lissenzephalie
Weibliche Mitglieder der in der Abbildung dargestellten Familie, welche eine Mutation im DCX-Gen tragen (Personen 1 und 7), entwickelten in jugendlichem Alter epileptische Anfälle, ohne jedoch weitere klinische Symptome zu zeigen. Die ebenfalls von der DCX-Mutation betroffenen männlichen Mitglieder der Familie (Personen 5, 6 und 10) hingegen litten bereits wenige Wochen nach ihrer Geburt unter schweren epileptischen Anfällen, waren stark in ihrer geistigen Entwicklung verzögert und körperlich schwer behindert.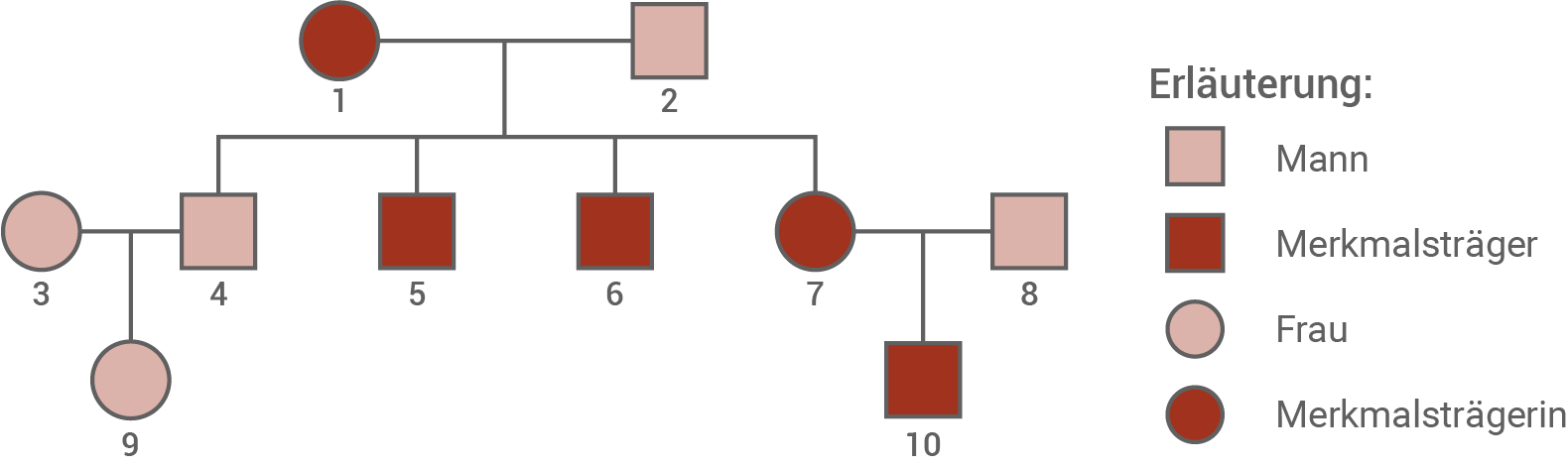
Deborah Morris-Rosendahl: Klinik, Genetik und Pathogenese der Lissenzephalien, Deutsches Ärzteblatt 2003, 100.A 1 269-1 282 (Heft 19), https://www.aerzteblatt.de/archiv/36794/Klinik-Genetik-und-Pathogenese-der-Lissenzephalien
Lisa Dey: Mutationssuche und deren Analyse in den LISI- und DCX-Genen bei Patienten mit Lissenzephalie, 2006, https://d-nb.info/981989217/34
Claudia Karl: Die Rolle des Doublecortin-Gens in neuronalen Vorläuferzellen während Migration und Neurogenese, 2004, https://epub.uni-regensburg.de/10328/1/Karl-C-2004.pdf
Material 5
Molekulargenetische Untersuchung eines Patienten mit Lissenzephalie
Bei einem Patienten mit klinisch diagnostizierter Lissenzephalie wurde das DCX-Gen molekulargenetisch untersucht. Das Genprodukt Doublecortin besteht aus 361 Aminosäuren.Abb. 5.1: Ausschnitte des nicht-codogenen DNA-Strangs des DCX-Gens
| 5´– ...GAC CTG ACG CGA TCT... – 3´ | gesunder Mann |
| 5´– ...GAC CTG AGG CGA TCT... – 3´ | Patient mit Lissenzephalie |
Abb. 5.2: Code-Sonne der mRNA

Lisa Dey: Mutationssuche und deren Analyse in den LISI-und DCX-Genen bei Patienten mit Lissenzephalie, 2006, https://d-nb.info/981989217/34
Material 6
Molekulargenetische Nachweismethoden
In einer weiteren molekulargenetischen Untersuchung des DCX-Gens des in Material 5 vorgestellten Patienten wurde zunächst ein Bereich von 469 Basenpaaren (bp) Länge aus dem kodierenden Bereich mittels PCR vervielfältigt. Die entstandenen PCR-Produkte wurden anschließend mit dem Restriktionsenzym DdeI behandelt. Die nicht mutierten Allele des DCX-Gens gesunder Personen weisen für dieses Enzym im untersuchten Bereich vier Schnittstellen auf. Die entstandenen Fragmente der PCR-Produkte wurden anschließend mittels Gelelektrophorese aufgetrennt.Abb. 6.1: Erkennungssequenz des Restriktionsenzyms Ddel
5´– ...CTNAG... – 3´3´– ...GANTC... – 5´ Erläuterung:
N: beliebige Base (Adenin, Cytosin, Guanin oder Thymin)
Abb. 6.2: Gelelektrophorese von PCR-Produkten des DCX-Gens nach Behandlung mit Ddel
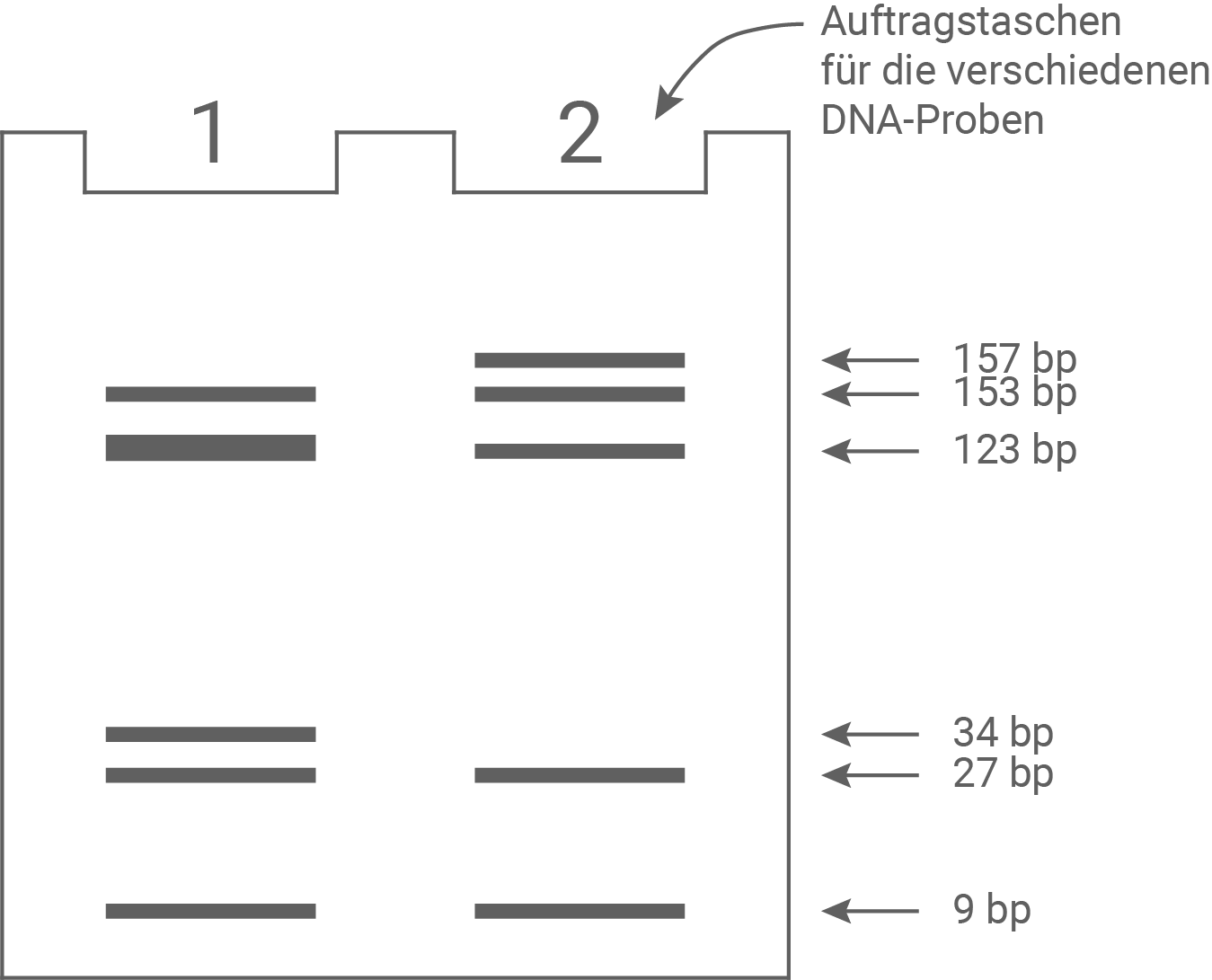
Erläuterung:
1: Patient mit Lissenzephalie
2: gesunder Mann
Dick dargestellte Banden symbolisieren eine doppelte DNA-Menge im Vergleich zu dünn dargestellten Banden.
1: Patient mit Lissenzephalie
2: gesunder Mann
Dick dargestellte Banden symbolisieren eine doppelte DNA-Menge im Vergleich zu dünn dargestellten Banden.
Lisa Dey: Mutationssuche und deren Analyse in den LISI- und DCX-Genen bei Patienten mit Lissenzephalie, 2006, https://d-nb.info/981989217/34
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?
1
Entstehung eines excitatorischen Potenzials:
Erreicht ein Aktionspotenzial die Präsynapse, so öffnen sich in deren Membran spannungsabhängige Calcium-Ionenkanäle, und es kommt zu einem Einstrom positiv geladener Calciumionen. Die dadurch hervorgerufene erhöhte Calcium-Ionenkonzentration in der Synapse bewirkt die Wanderung der mit dem Transmittermolekül Acetylcholin gefüllten synaptischen Bläschen. Diese bewegen sich in Richtung der präsynaptischen Membran, wo sie schließlich mit ihr verschmelzen. Durch Exocytose wird der Transmitterstoff in den synaptischen Spalt entlassen, und gelangt durch Diffusion bis zur postsynaptischen Membran. In der postsynaptischen Membran befinden sich ligandengesteuerte Natrium-Ionenkanäle. Acetylcholin ist in der Lage, an diese zu binden, wodurch die Öffnung der Natrium-Ionenkanäle induziert wird. Positiv geladene Natriumionen strömen in die Zelle ein, und lösen so eine Depolarisation der postsynaptischen Membran aus. Es entsteht ein EPSP (exzitatorisches postsynaptisches Potenzial).
Entstehung eines inhibitorischen Potenzials:
Inhibitorische Potenziale entstehen an der Membran hemmender Synapsen. Binden die Transmittermoleküle an die postsynaptische Membran, so werden Kalium-Ionenkanäle oder Chlorid-Ionenkanäle geöffnet. Auf diese Weise strömen entweder Kalium-Ionen aus der Zelle hinaus, oder Chlorid-Ionen in die Zelle hinein. Der negative Ladungsüberschuss in der Zelle löst eine Hyperpolarisation aus. Das wird als inhibitorisches Potenzial bezeichnet.
Definition der Begriffe zeitliche und räumliche Summation:
- Unter dem Begriff zeitliche Summation versteht man die Verrechnung aller EPSP oder IPSP, die innerhalb eines bestimmten Zeitraums ausgehend von einer einzelnen Synapse in der Nervenzelle ankommen.
- Unter dem Begriff räumliche Summation versteht man die Verrechnung aller EPSP oder IPSP, die zeitgleich, ausgehend von verschiedenen Synapsen, in der Nervenzelle ankommen.
2
Erklärung der Messergebnisse an den Messstellen 1 und 2:
Bei einer künstlichen Reizung des Motoneurons wird durch die Axonkollerate auch das Renshaw-Neuron gereizt. Innerhalb kurzer Zeitabstände bildet es viele Aktionspotenziale, die an Messstelle 1 gemessen werden können. Das Renshaw-Neuron ist synaptisch mit dem Soma des Motoneurons verbunden. Am Zellkörper des Motoneurons kann eine Hyperpolarisation gemessen werden. Dies ist die Reaktion auf den vom Renshaw-Neuron ausgeschütteten Transmitterstoff Glycin. Das Renshaw-Neuron wirkt damit hemmend auf das Motoneuron.
Bedeutung von Renshaw-Neuronen für die Steuerung der Muskelkontraktion bei der Aktivierung von Motoneuronen durch absteigende Nervenbahnen:
Das Motoneuron wird durch die absteigenden Nervenbahnen erregt. Es entsteht ein EPSP am Soma und im Axon werden Aktionspotenziale gebildet. Diese lösen an den motorischen Endplatten Potenziale aus, was eine Kontraktion des Muskels zur Folge hat. Durch die Reizweiterleitung wird aber auch das Renshaw-Neuron angeregt, welches am Soma des Motoneurons ein IPSP generiert. Am Soma findet nun eine räumliche Summation der Erregungen statt. Das von den absteigenden Nervenbahnen kommende EPSP wird mit dem IPSP des Renshaw-Neurons verrechnet. Infolgedessen wird die Frequenz der Aktionspotenziale verringert, wodurch die Muskelzellen der motorischen Endplatte und damit der gesamte Muskel nicht so stark kontrahiert. Die Funktion der Renshaw-Neuronen liegt also darin, die Stärke der von dem Motoneuron abgegebenen Erregung und damit die Muskelaktivität zu regulieren.
3
Analyse der Untersuchungsergebnisse in Material 2:
Im Experiment wurden für Epilepsie anfällige Ratten einem akustischen Reiz ausgesetzt, der unter normalen Umständen einen epileptischen Anfall auslösen kann. Allerdings wurde ihnen zuvor der Wirkstoff Topiramat verabreicht. Es wurde beobachtet, ob die Ratten für den Anfall typische Symptome wie eine Rennphase gefolgt von einer Muskelstarre zeigten. Alle acht Ratten der Kontrollgruppe, denen kein Topiramat verabreicht wurde, zeigten diese Symptome. Auch die Ratten, denen 10 mg Topiramat pro kg Körpergewicht verabreicht wurde, verhielten sich so. Ab einer Konzentration von 30 mg Topiramat zeigten zwar immer noch alle acht Ratten die Rennphase, jedoch litt nur noch eine Ratte an der anschließenden Starre. Diese Situation stellte sich auch für eine Topiramatkonzentration von 60 mg ein. Im Experiment konnte gezeigt werden, dass der Wirkstoff Topiramat ab einer Konzentration von 30 mg pro kg Körpergewicht den Zustand der Starre bei Ratten mit einem epileptischen Anfall mit einer hohen Wahrscheinlichkeit verhindern kann. Die Rennphase kann bei keiner der verabreichten Konzentrationen verhindert werden.
4
Verschiedene Wirkorte des Antiepileptikums Topiramat an Glutamat führenden Synapsen:
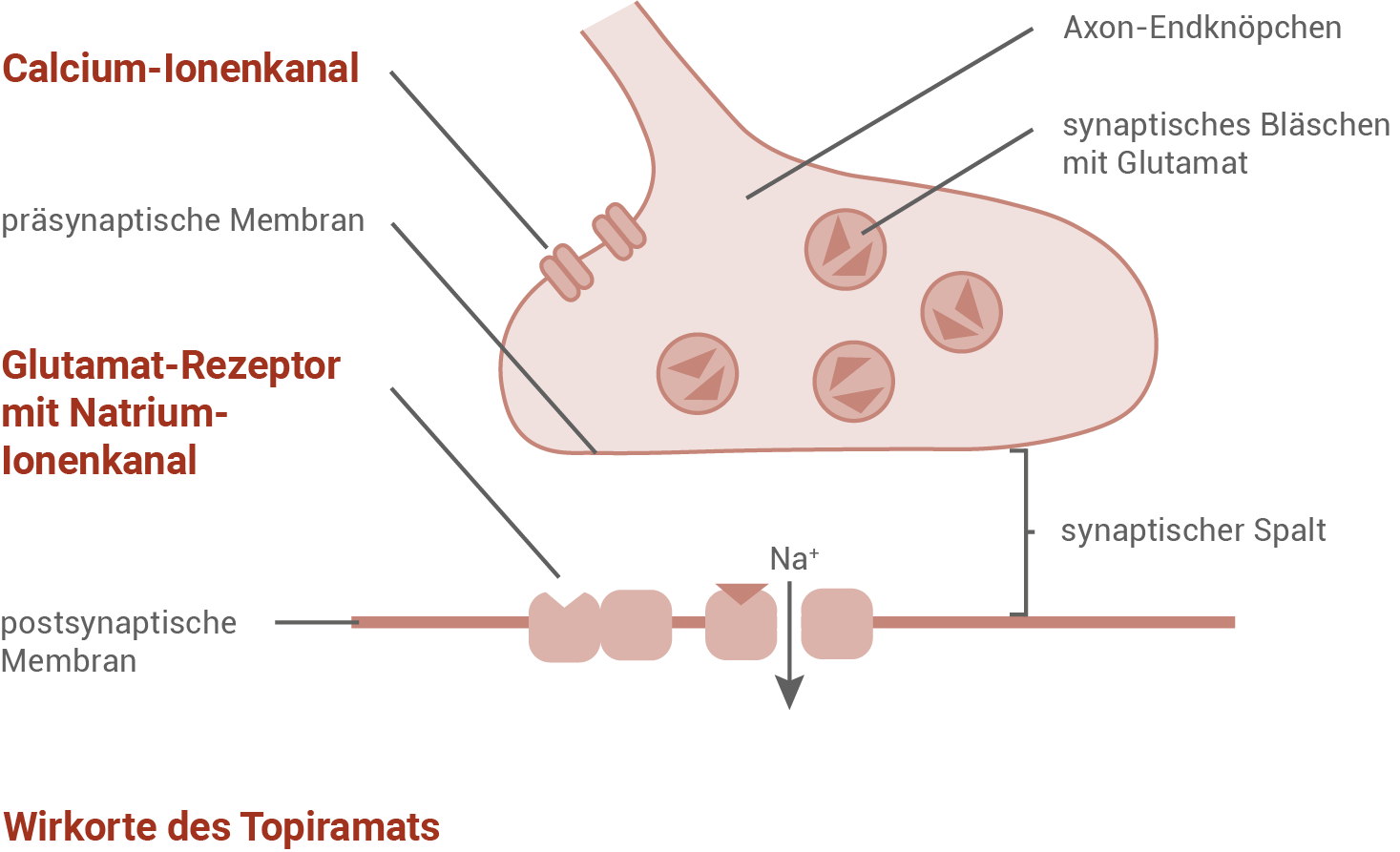 Einfluss von Topiramat auf die Erregungsübertragung der Synapsen:
Topiramat blockiert die spannungsabhängigen Calcium-Ionenkanäle in der Präsynapse, wodurch keine Calciumionen in die Zelle gelangen können. Durch den blockierten Calcium-Ioneneinstrom verschmelzen die synaptischen Bläschen mit dem eingeschlossenen Transmitter nicht mit der präsynaptischen Membran, und werden nicht in den synaptischen Spalt entlassen. Topiramat bindet zudem an die Glutamat-Bindestellen der Rezeptoren in der postsynaptischen Membran, ohne diese zu öffnen. Da die Rezeptoren nun blockiert sind, kann Glutamat nicht mehr an sie binden, der Natriumeinstrom unterbleibt, und es wird kein EPSP gebildet. Bei beiden Wirkmechanismen ist eine Einschränkung der Erregungsweiterleitung die Folge.
Einfluss von Topiramat auf die Erregungsübertragung der Synapsen:
Topiramat blockiert die spannungsabhängigen Calcium-Ionenkanäle in der Präsynapse, wodurch keine Calciumionen in die Zelle gelangen können. Durch den blockierten Calcium-Ioneneinstrom verschmelzen die synaptischen Bläschen mit dem eingeschlossenen Transmitter nicht mit der präsynaptischen Membran, und werden nicht in den synaptischen Spalt entlassen. Topiramat bindet zudem an die Glutamat-Bindestellen der Rezeptoren in der postsynaptischen Membran, ohne diese zu öffnen. Da die Rezeptoren nun blockiert sind, kann Glutamat nicht mehr an sie binden, der Natriumeinstrom unterbleibt, und es wird kein EPSP gebildet. Bei beiden Wirkmechanismen ist eine Einschränkung der Erregungsweiterleitung die Folge.
5
Ablauf einer Polymerase-Kettenreaktion:
- Denaturierung: Die Doppelhelix wird bei einer Temperatur von ca. 95 °C in ihre beiden Einzelstränge aufgespalten. Das liegt daran, dass bei dieser Temperatur die Wasserstoffbrücken zwischen den einzelnen Basenpaaren aufgelöst werden.
- Hybridisierung: Hier werden bei 55 bis 60 °C Primer komplementär an die zu vervielfältigenden Abschnitte angelagert.
- Polymerisation: Die sogenannte Taq-Polymerase synthetisiert bei 72 °C ausgehend von dem 3'-Ende des DNA-Primers einen zum Matrizenstrang komplementären DNA-Strang.
- Dieser Zyklus wird so oft wiederholt, bis die gewünschte Menge des zu replizierenden DNA-Abschnittes entstanden ist.
6
Mögliche Vererbungsmodi von Lissenzephalie anhand des Stammbaums aus Material 4:
- Da sowohl Frauen als auch Männer betroffen sind, ist davon auszugehen, dass der Vererbungsmodus nicht Y-chromosomal abläuft.
- Ein X-chromosomal-rezessiver Erbgang ist nicht möglich, da aus der Verbindung einer betroffenen Mutter (Person 1) und eines gesunden Vaters (Person 2) ein gesunder Sohn (Person 4), zwei betroffene Söhne (Personen 5 und 6) und eine betroffene Tochter (Person 7) hervorgehen. Würde ein X-chromosomaler Erbgang vorliegen, dürfte die Tochter aufgrund des nicht mutierten väterlichen X-Chromosoms nicht erkranken, alle Söhne des Paares müssten jedoch erkrankt sein.
- Denkbar wäre eine autosomal-rezessive Vererbung des mutierten DCA-Allels. Dabei wären die beiden phänotypisch gesunden Väter (Personen 2 und 8) jeweils heterozygote Träger des mutierten Allels. Wird dieser Erbgang angenommen, wären die Mütter (Personen 1 und 7) homozygot erkrankt und würden das mutierte DCX-Gen an ihre Kinder vererben, sodass aus der Verbindung mit einem heterozygot gesunden Vater sowohl heterozygot gesunde als auch homozygot kranke Söhne und Töchter hervorgehen können.
- Eine autosomal-dominante sowie eine X-chromosomal-dominante Vererbung wären ebenfalls möglich. Dann wäre die Mutter (Person 1) in der ersten Generation Konduktorin des mutierten Allels und drei ihrer Kinder (Personen 5, 6 und 7) hätten dieses Allel erhalten und wären als heterozygote Träger des dominanten Allels phänotypisch krank, während der eine Sohn (Person 4) von seiner Mutter das unveränderte Allel erhalten hätte und dementsprechend homozygot gesund wäre.
7
Aminosäuresequenz des DNA-Ausschnittes des DCX-Gens einer an Lissenzephalie erkrankten Person:
Asp – Leu – Arg – Arg – Ser
Bestimmung der Mutationsform und Erklärung möglicher Folgen für die Gehirnentwicklung:
Es liegt eine Punktmutation vor. Dabei ist die Base Cytosin gegen die Base Guanin ausgetauscht worden. Damit handelt es sich um eine Basensubstitution. In Konsequenz wird bei der Translation die Aminosäure Arginin anstelle von Threonin in die Polypeptidkette eingebaut. Die Missense-Mutation hat zur Folge, dass das synthetisierte Protein Doublecortin nicht voll funktionsfähig ist. Durch die Mutation kann die Bindefähigkeit des Proteins an die Mikrotubuli beeinträchtigt sein. Das wachsende Cytoskelett könnte dadurch in seiner Stabilität limitiert sein. Die Einwanderung und korrekte Positionierung junger Neuronen in die Großhirnrinde während der Gehirnentwicklung kann dadurch nicht ungehindert ablaufen.
8
Unterschiede in den Ergebnissen der Gelelektrophorese im Vergleich:
Die Gelelektrophorese zeigt die mit dem Restriktionsenzym DdeI behandelten PCR-Produkte des DCX-Gens eines Patienten mit Lissenzephalie (Person 1) und eines gesunden Mannes (Person 2). Beide Personen weisen gleiche Banden mit einer Länge von 9, 27 und 153 Basenpaaren Länge auf. Die erkrankte Person besitzt eine zusätzliche Bande bei 34 bp, und die doppelte DNA-Menge bei 123 bp. Der erkrankten Person fehlt die Bande bei 157 bp.
Deutung des Befundes bei dem Lissenzephalie-Patienten:
Das Restriktionsenzym DdeI scheidet das PCR-Produkt des nicht mutierten DCX-Gens an vier Stellen, wodurch beim gesunden Mann fünf Fragmente entstehen. Bei dem Patienten mit Lissenzephalie ergibt sich durch die Basenpaarsubstitution eine weitere Schnittstelle für das Restriktionsenzym. Das Fragment der Länge 157 bp wird dadurch in zwei Fragmente der Länge 123 bp und 34 bp gespalten.