Vorschlag B: Muskeldystrophien
Genetik verschiedener Formen der Muskeldystrophie
Muskeldystrophie aus neurobiologischer Sicht
Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?Material 1
Arten der Muskeldystrophie
Abbildung 1.1: Stammbaum einer von BMD betroffenen Familie
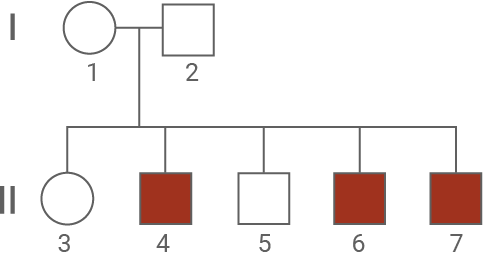
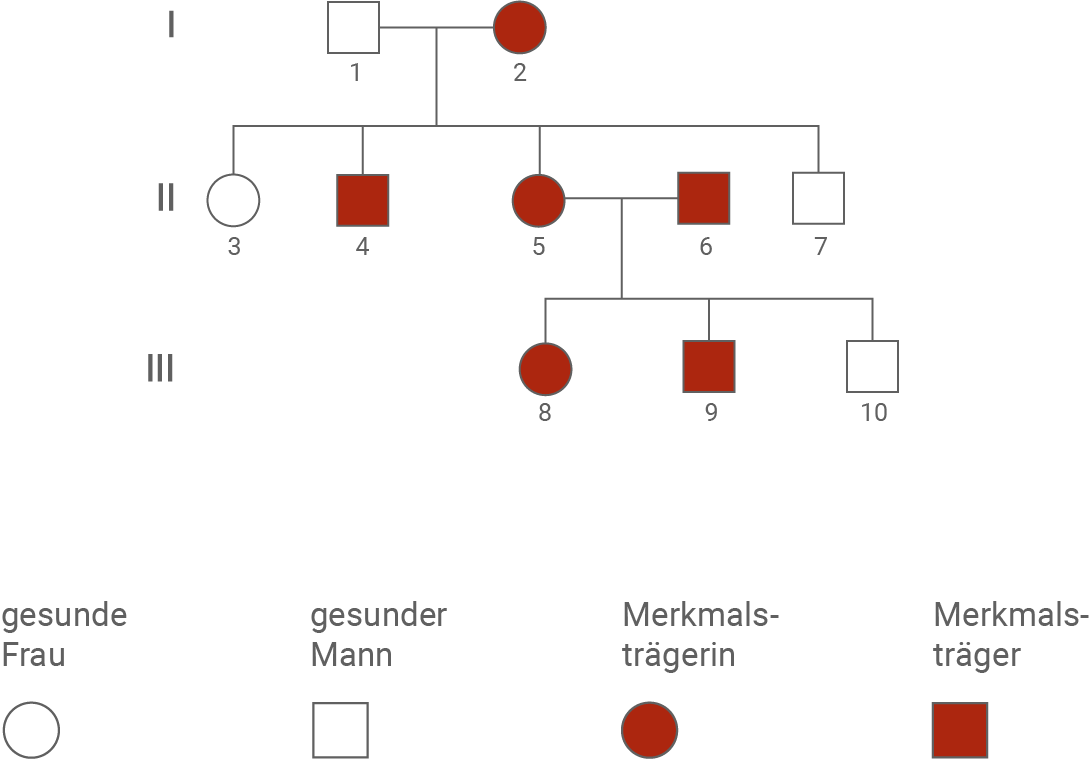
Abbildung 1.2: Stammbaum einer von FSHD betroffenen Familie
Material 2
Molekulargenetische Untersuchungsergebnisse zur Duchenne- und Becker-Muskeldystrophie
Ausschnitt aus dem codogenen DNA-Strang des Dystrophin-Gens:
| Codon Nummer | gesunde Person A | an Duchenne-Muskeldystrophie (DMD) erkrankte Person B: |
|---|---|---|
| ... 2482...................2485 ... | 3′... GAA TGG CTG ACC ...5′ | 3′... GAA GGC TGA CCG ...5′ |
| Codon Nummer | gesunde Person C | an Becker-Muskeldystrophie (BMD) erkrankte Person D: |
|---|---|---|
| ... 2482...................2485 ... | 3′... TTG GCG CCA CTG ...5′ | 3′... TTG GTG CCA CTG ...5′ |
Material 3
Funktion von Muskelzellen bei Personen ohne und mit DMD
Abbildung 3.1: Ausschnitt einer Muskelzelle bei gesunden Personen im entspannten Zustand
DG, P1, P2 Proteinkomplexe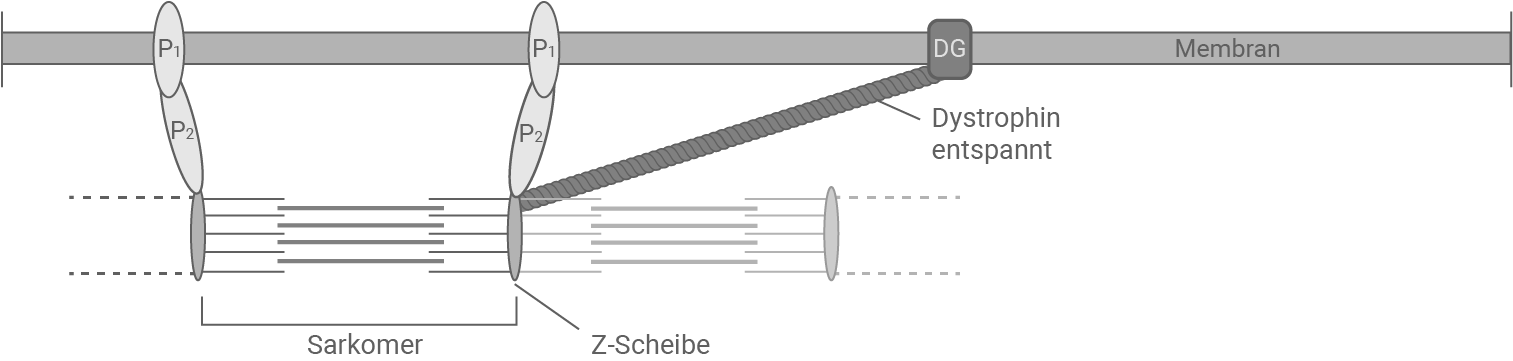
Abbildung 3.2: Ausschnitt einer Muskelzelle bei gesunden Personen bei vollständiger Sarkomer-Aktivierung
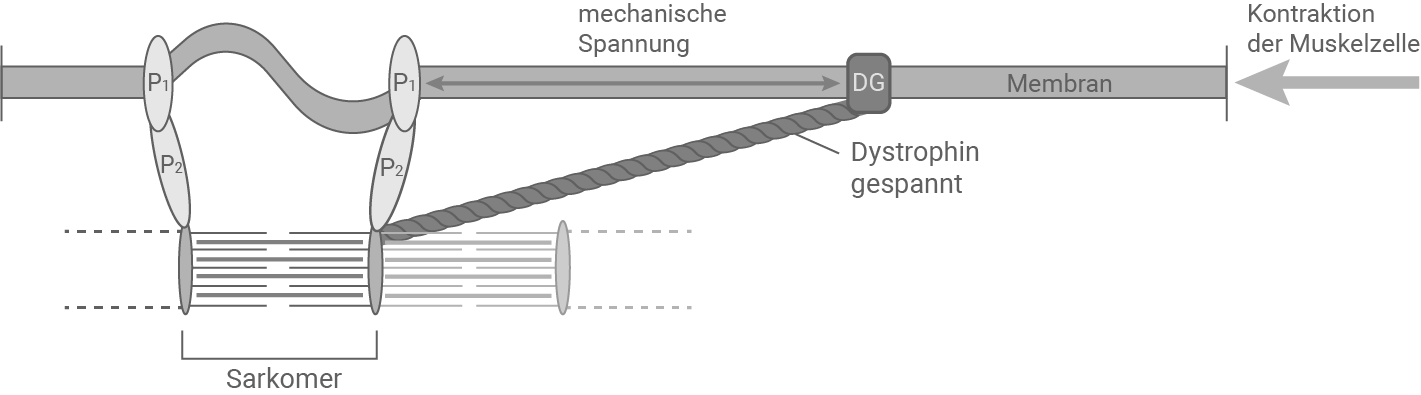
Abbildung 3.3: Ausschnitt einer Muskelzelle bei Personen mit DMD zu Beginn der Sarkomer-Aktivierung
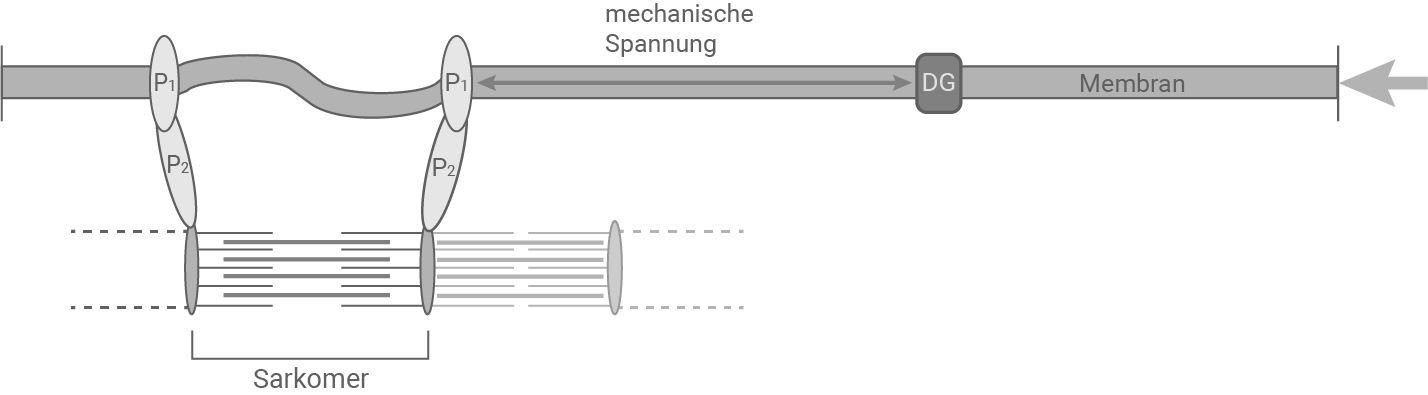
Abbildung 3.4: Ausschnitt einer Muskelzelle bei Personen mit DMD bei vollständiger Sarkomer-Aktivierung
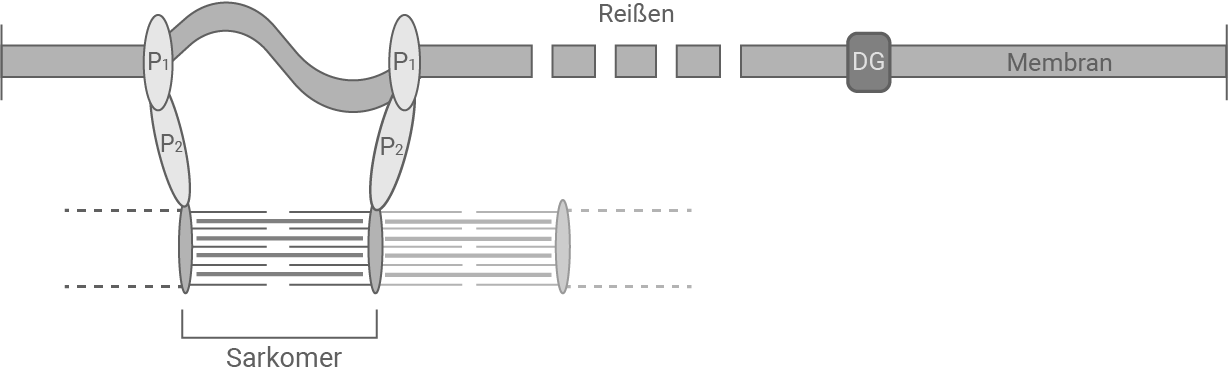
Material 4
Erregungsübertragung an einer Glutamat führenden Synapse
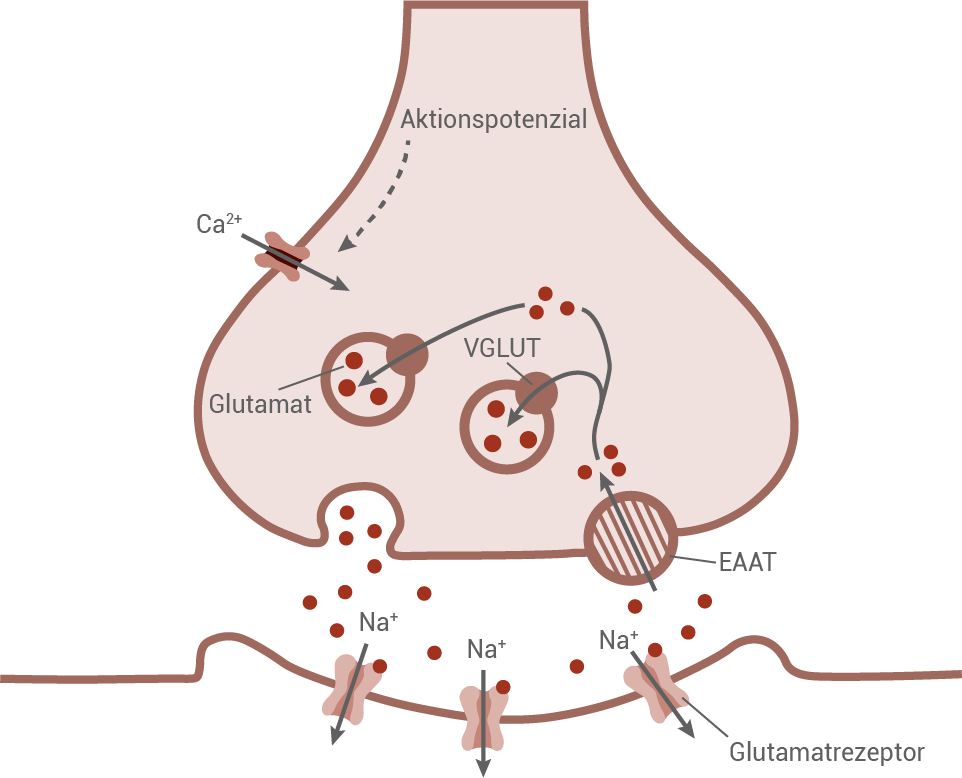
Material 5
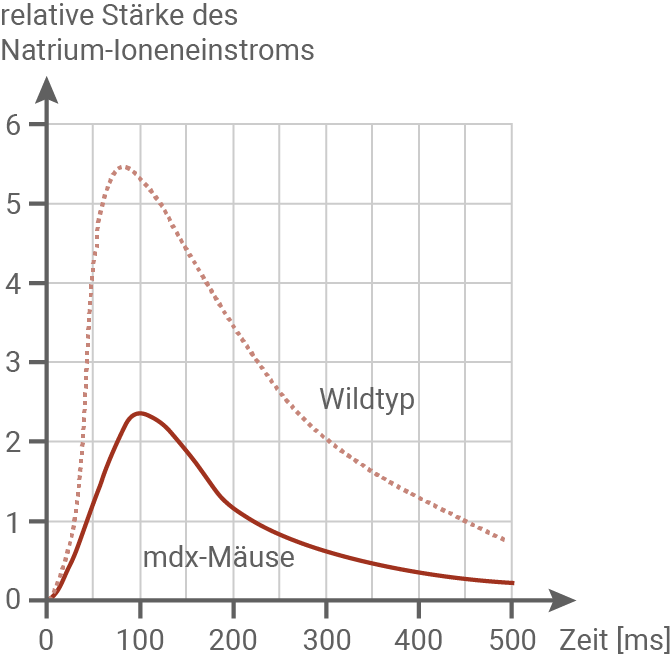
Einfluss von Dystrophinmangel auf die synaptische Übertragung
Ionenstrom durch die postsynaptische Membran
Material 6
Verhalten von mdx-Mäusen
Abbildung 6.1: Versuchsaufbau des Morris-Wasserlabyrinths
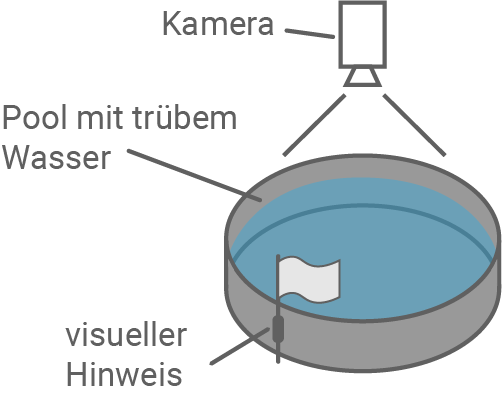
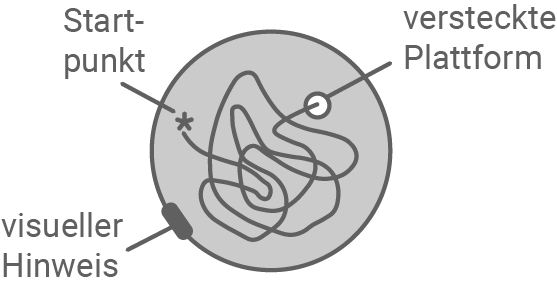
Abbildung 6.2: Orientierung im Morris-Wasserlabyrinth
Code-Sonne der mRNA

Weiter lernen mit SchulLV-PLUS!
monatlich kündbarSchulLV-PLUS-Vorteile im ÜberblickDu hast bereits einen Account?- Initiation: Das Ribosom fährt an der mRNA-Sequenz entlang, bis es auf das Startcodon trifft. Das Start-Codon befindet sich nun an der sogenannten P-Stelle des Ribosoms. Daran bindet eine passende tRNA, die ein Anti-Codon besitzt, das komplementär zum Codon auf der mRNA ist. Jedes Anti-Codon steht dabei für eine bestimmte Aminosäure. Die tRNA trägt diese Aminosäure am oberen Ende. Nun lagert sich eine weitere tRNA an die freie A-Stelle an.
- Elongation: Die Aminosäure der tRNA in der P-Stelle hängt sich daraufhin an die Aminosäure der tRNA in der A-Stelle an. Das Ribosom wandert ein Triplett weiter. Damit befindet sich die erste tRNA an der E-Stelle und löst sich vom Ribosom. In die freie A-Stelle kann sich jetzt eine neue tRNA anlagern. Die tRNA mit der wachsenden Aminosäurekette befindet sich damit in der P-Stelle. Von dort aus können die Aminosäuren wieder zur tRNA an der A-Stelle wandern. Dieser Prozess wiederholt sich immer wieder, und die Polypeptidkette wächst.
- Termination: Trifft das Ribosom auf ein Stopp-Codon, löst sich die Polypeptidkette von der tRNA. Sie wird ins Cytoplasma freigegeben, und faltet sich dort in ihre dreidimensionale Protein-Struktur.
- Der Stammbaum zeigt, dass zwei phänotypisch nicht betroffene Eltern erkrankte Kinder haben. Dieses Muster kann nur bei einem rezessiven Erbgang auftreten. Wäre der Erbgang dominant, so müssten mindestens ein Elternteil auch betroffen sein, damit die Krankheit bei den Kindern ausgeprägt wird.
- Es sind nur Männer von der Krankheit betroffen. Damit ist der Erbgang X-chromosomal. Die Mutter (1) muss Konduktorin sein, und ihr betroffenes X-Chromosom an die betroffenen Söhne weitergeben. Der Vater muss gesund sein, da er nur ein X-Chromosom besitzt, und die Krankheit auch ausprägen müsste, wenn dieses X-Chromosom betroffen wäre.
- Die Wahrscheinlichkeit, dass eine gesunde Tochter geboren wird, liegt bei 100 %, da sie maximal ein defektes X-Chromosom der Mutter erbt. Das andere X-Chromosom vom Vater ist nicht betroffen. Allerdings besteht eine 50-prozentige Wahrscheinlichkeit, dass die Tochter Konduktorin ist. Dabei besitzt sie ein betroffenes X-Chromosom, prägt die Krankheit aber selbst nicht aus.
- Die Wahrscheinlichkeit für einen gesunden Sohn beträgt 50 %, da er entweder gesunde oder betroffene X-Chromosom der Mutter erbt. Da er nur ein X-Chromosom erbt, kann ein erkranktes Chromosom nicht mit einem gesunden kompensiert werden.
- Autosomal-dominanter Erbgang:
Person 8: Aa oder AA
Person 9: Aa oder AAEs ist A: betroffenes Allel; a: gesundes Allel - Gonosomal-dominanter Erbgang:
Person 8: XX oder Xx
Person 9: XyEs ist X: betroffenes X-Chromosom; x: gesundes X-Chromosom; Y: betroffenes Y-Chromosom; y: gesundes Y-Chromosom
- Person A (gesund):
mRNA-Sequenz: CUU ACC GAC UGG
Aminosäuresequenz: Leu-Thr-Asp-Trp - Person B (DMD-Patient): mRNA-Sequenz: CUU CCG ACU GGC
Aminosäuresequenz: Leu-Pro-Thr-Gly - Person C (gesund):
mRNA-Sequenz: AAC CGC GGU GAC
Aminosäuresequenz: Asn-Arg-Gly-Asp - Person D (BMD-Patient):
mRNA-Sequenz: AAC CAC GGU GAC
Aminosäuresequenz: Asn-His-Gly-Asp
- Person B: Es handelt sich um eine Deletion einer Base, die eine Leserastermutation hervorruft. Bei der gesunden Person A befindet sich an der ersten Stelle von Triplett Nummer 2483 die Base Thymin. Bei der erkrankten Person B fehlt sie, und das Leseraster verschiebt sich um eine Position nach links. Ab dieser Stelle werden daher andere Aminosäuren in die Polypeptidkette eingebaut, als bei der gesunden Person. Da verschiedene Aminosäuren unterschiedliche Wechselwirkungen aufweisen, entsteht ein komplett anderes Protein, als bei gesunden Personen. Vermutlich kann das mutierte Dystrophin-Protein seine ursprüngliche Funktion nicht mehr ausführen.
- Person D: Hier liegt eine Basensubstitution vor, die zu einer Missense-Mutation führt. In Triplett Nummer 1745 wurde durch eine Punktmutation die Base Cytosin mit Thymin substituiert. Infolgedessen codiert dieses Triplett nicht mehr für Arg, sondern für His. Da die Mutation nut ein Triplett betrifft, bleiben die anderen Aminosäuren unverändert. Die Mutation führt dennoch zu einer Veränderung der Struktur und damit zur Beeinträchtigung der Proteinfunktion.
Bei einer Person mit DMD fehlt das Dystrophin-Protein. Während der Kontraktion ist die mechanische Spannung der Membran höher, da sie nicht zum Teil von Dystrophin getragen wird. Eine vollständige Kontraktion belastet die Membran so sehr, dass sie reißt.
Die Folge für die betroffenen Patienten ist eine fortschreitende Muskeldegeneration, da bei jeder Muskelkontraktion Muskelzellen durch das Reißen der Membran absterben. Ohne die Kontraktion von Muskeln können diese auch nicht aufgebaut werden. In Kombination mit dem Abbau der abgestorbenen Zellen führt das zu einem Verlust der Muskelmasse.
In dem Morris-Wasserlabyrinth-Versuch wurden die Orientierungsfähigkeiten von Wildtyp-Mäusen (mit DP-140) und mdx-Mäusen (ohne DP-140) untersucht. Ziel war es, die Auswirkungen des fehlenden DP-140 auf kognitive Leistungen und Gedächtnis zu bewerten.
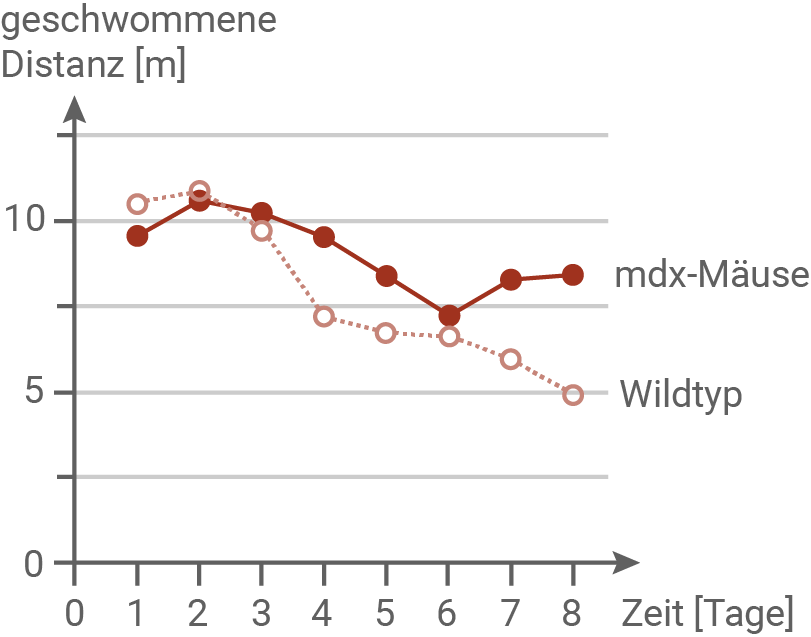
Abbildung 6.2 zeigt, dass Wildtyp-Mäuse die versteckte Plattform schneller finden und sich besser an deren Position erinnern können. Dabei ist die geschwommene Distanz in Metern auf der y-Achse und die Tage an denen das Experiment durchgeführt wurde auf der x-Achse dargestellt. Zu Beginn des Experiments gleichen sie die zurückgelegten Distanzen nahezu. Ab dem vierten Tag lassen sich jedoch stärkere Unterschiede in den beiden Gruppen beobachten. Während bei den Wildtypmäusen die Distanz über den gesamten Zeitraum abnimmt, ist die Abnahme bei den mdx-Mäusen zunächst schwächer. Ab Tag sechs steigt die Distanz sogar wieder. Es ist davon auszugehen, dass Wildtyp-Mäuse recht schnell lernten, den visuellen Hinweis am Beckenrand als Orientierungshilfe zu nutzen. Dadurch fanden sie die Platform mit zunehmender Zeit immer schneller. Mäuse ohne DP-140 benötigen mehr Zeit, um die Plattform zu finden, und zeigen ein weniger zielgerichtetes Schwimmverhalten. Das deutet auf eine beeinträchtigte räumliche Orientierung und Gedächtnisleistung hin.
Interpretation der Ergebnisse:Das fehlende DP-140 scheint die Funktion der Amygdala und des Hippocampus zu beeinflussen, die für Gedächtnis und räumliche Orientierung entscheidend sind. Ohne DP-140 ist die synaptische Übertragung von Glutamat gestört, was zu kognitiven Defiziten durch eine schlechtere Signalweiterleitung führt.
Die Ergebnisse unterstreichen, dass Dystrophin nicht nur für die Stabilität der Muskeln, sondern auch für die neuronale Funktion essenziell ist. Das erklärt, warum Patienten mit DMD häufig auch Lern- und Gedächtnisschwierigkeiten zeigen.